



Portola Pharmaceuticals: Initiating Coverage with a Buy (PTLA, $40.05)
��Overview of Report
I am initiating coverage of Portola Pharmaceuticals with a Buy and a 2021 price target of $426.This is an extensive initial report on Portola Pharmaceuticals that is organized into five parts:
- Investment Overview for Portola is a summary of the key investment issues. An investor only needs to read this section in order to gain an understanding of the investment issues. The remainder of the report is for those who want to dig into complex details
- Basic Biology Background on Coagulation discusses the biology involved in coagulation and anticoagulant drugs used to treat coagulation diseases. This is the space that Portola’s products are operating in.
- What Is the Role of Betrixaban is a detailed summary of the disease target, the clinical trial program and sales projections for betrixaban.
- Addressing the Medical Need for Antidotes to Factor Xa Inhibition is a detailed summary of the disease target , the clinical trial program and sales projections for AndexXa (andexamet)
- Five Year Sales Projections for Betrixaban and AndexXa
Investment Overview for Portola
Two Potential Blockbuster Drugs are Close to Approval
Portola has two drugs in late stage development that I believe are likely to be approved in 2017 or early 2018 in both the US and Europe. Betrixaban is an anticoagulant drug that is an oral factor Xa inhibitor. It has the same mechanism of action as two blockbuster drugs: Bristol-Myers Squibb/ Pfizer’s Eliquis ($3.3 billion of sales in 2016) and Johnson & Johnson/ Bayer’s Xarelto ($5.0 billion of sales in 2016). However, its distinctive drug properties differentiate it from these two drugs. It could receive approval for extended duration, anticoagulant treatment of acute medically ill patients at its June 24, 2017 PDUFA date. Importantly, phase 3 trials for Eliquis and Xarelto failed so that this will be the only drug approved in this indication. I estimate that by 2022, US sales of betrixaban could reach $925 million and rest of world sales (primarily Europe) could reach $400 million.
The second product, AndexXa (andexamet) is labeled by the FDA as a breakthrough medicine that addresses a critical unmet medical need for a drug that can reverse the anticoagulant activity of drugs like Xarelto and Eliquis. It is the only such drug close to commercialization. The mechanism of action used by Xarelto and Eliquis is the inhibition of the factor Xa protein which is an integral element in the clotting cascade; the intricate biological process that creates a blood clot. Reducing the ability of the blood to clot can prevent life threatening strokes and ischemia. However, it also carries an offsetting major risk because they can cause spontaneous bleeds or exacerbate bleeding stemming from emergency surgery or trauma. These events can be life threatening. I estimate that by 2022, US sales could reach $560 million and rest of world sales of $350 million.
Patent Status
The betrixaban patent portfolio includes 22 issued U.S. patents and 4 U.S. patent applications covering the composition of and methods of making and using betrixaban or its analogs. The U.S. issued patents relating to the composition of matter of betrixaban are due to expire in September 2020. Under the provisions of the Hatch-Waxman Act, betrixaban will almost certainly receive a five year extension to September 2025. This can be extended by another six months to March 2026 if studies of the drug in children are conducted pursuant to a request from the FDA. The Company has not provided sufficient information to judge whether other patents could extend this period of exclusivity.
The andexanet alfa portfolio is wholly owned by Portola. It includes 10 issued U.S. patents and 13 U.S. patent applications covering the composition of matter and methods of making and using andexanet or its analogs. The last to expire of the U.S. patents will last until July 2030.
International patent protection is comparable to the US for both products.
Betrixaban is Likely to be Approved in the US and Europe by Yearend
The progression through clinical trials and into the registration process has seen considerable drama for both drugs. On March 24, 2016, Portola announced that betrixaban had narrowly missed the primary endpoint of the pivotal APEX phase 3 trial as it achieved a p value of 0.054 slightly more than the p-value of 0.05 that is the (arbitrary) hurdle that the FDA has established to determine if a clinical trials is a success. However, a closer analysis persuades me (and many others) that the trial should and will be considered a success. This is a bit complicated to explain succinctly and later in this report I go into more detail, but here is a quick summary.
The statistical design divided the trial into three cohorts. The first cohort of 3,870 patients was comprised of patients who were felt to be at most risk as determined by d-dimer (a biomarker) levels. Cohort 2 was based on 5,735 patients; it included patients from cohort 1 plus additional patients felt to be at high risk because of age. Cohort 3 included 6,286 patients including those from cohort 2 and additional lower risk patients. The statistical plan designated the primary outcome in cohort 1 as being the determinant for success in the trial and results from cohort 2 and 3 would be only informational if the primary endpoint in cohort 1 was not met. The thinking was that the more severely ill patients of cohort 1 would benefit most from the therapy and the less severely ill patients of cohorts 2 and 3 were less likely to.
Clinical trials can sometimes confound drug developers and that was certainly the case here. The p value in cohort 1 was 0.054. However the p value in the cohort 2 group that included cohort 1 patients and additional patients felt to be at somewhat lesser risk produced a strong p-value of 0.03 and the still larger cohort 3 group including even more low risk patients produced a p value of 0.006. These results suggest that patients judged to be at higher risk received benefit but did less well and this is hard to explain; perhaps it was just a chance outcome. Had the trial prospectively defined the measure of success as the outcome in either cohort 2 or 3, the trial would have been a smashing success.
Another issue to consider is that one patient in cohort 1 had been randomized to receive enoxaparin, the control treatment in the study. On day 35, they had a positive ultrasound indicating deep vein thrombosis, but did not demonstrate symptoms until day 37. There was another positive ultrasound on day 40. The contract research organization concluded that this patient did not have an event during the period that was proscribed in the trial design. Portola disagreed with this classification and was supported by independent data analysis committees at Harvard and Duke. They believed that a positive ultrasound should classify as an event regardless of whether the patient was exhibiting symptoms. If this patient was reclassified as having an event the relative risk reduction would be increased from 19.4% to 19.8% and the p value for the trial would be 0.048 and statistically significant versus the p=0.054 that the CRO reported.
It is also important to point out that the safety profile for betrixaban was very positive as the incidence of major bleeding in all patients was no different than the control group that received enoxaparin. Xarelto in its major phase 3 MAGELLAN trial in this same indication met its efficacy endpoint but the incidence of major bleeding tipped the benefit risk ratio unfavorably and the trial was judged a failure. Similarly the large ADOPT phase 3 trial of Eliquis in this indication showed adequate safety, but the efficacy endpoint was not met. Betrixaban appears to be effective where Xarelto and Eliquis failed.
As I look at the totality of evidence from APEX it seems very clear that this drug is effective and safe in this indication and I think that most investors would agree. The FDA has been known for maintaining statistical purity and there was and remains a concern that the FDA might require another phase 3 trial. However, we are living in the age of Trump and the FDA is being pushed into injecting a measure of common sense into a decision like this as opposed to just sticking to the rigid guide of p≤ .05. Remember that a p value of 0.05 means that there is only a 5% chance that the outcome occurred by chance and a p value of 0.54 means that there is only a 5.4% chance.
Subsequent actions by the FDA indicate that the agency is likely to approve the drug. Portola submitted the NDA on October 15, 2016 and the FDA accepted the filing on December 23, 2016. The drug was assigned priority review which means that the agency considers this an important advance and a PDUFA data of June 24, 2017 was set. Then on February 8, 2017, the agency informed Portola that there would be no advisory committee called to review the NDA. This indicates that the FDA doesn’t have any concerns for which it needs guidance from key opinion leaders. I believe that betrixaban will be approved on June 24. 2017.
AndexXa Approval in the US in Late 2017 is Likely
The BLA for AndexXa was filed on January 4, 2016. The FDA had previously designated it as a breakthrough drug and a priority review was assigned with a PDUFA date of August 19, 2016. Disappointingly, the FDA issued a complete response letter on that PDUFA date. However, the issues did not relate to clinical data but to manufacturing issues stemming from documenting quality in the manufacturing process. Portola has said that it believes that it can answer agency concerns without any new trials and plans to re-submit the BLA in 2Q, 2017. This could lead to approval in 4Q, 2017. Manufacturing issues can be tricky, but they still are of less concern than problems with clinical data. If Portola is correct, the product should be approved in late 2017 and launched in early 2018.
European Approvals
The EMA validated Portola’s marketing authorization application (MAA) for betrixaban in December 2016 and assigned a standard review. PTLA expects to receive questions from the Committee for Medicinal Products for Human Use (CHMP) and anticipates an approval towards the end of 2017. The MAA for AndexXa was validated in August of 2016. Portola expects an opinion from CHMP in December of 2017 with approval anticipated in the first quarter of 2018.
Commercialization Plans
PTLA is prepared to launch betrixaban into the US hospital market in August 2017, if it is approved. Beginning in early 2016, they began to hire strategic marketers, sales managers, managed care relationship personnel and medical affairs specialists. They plan to contingently hire between now and the August PDUFA date hospital sales reps with extensive experience in the field of thrombosis in the hospital setting. This team would then be expanded as adoption for betrixaban expands and AndexXa is approved. They might expand to 125 to 150 reps in the US by the end of 2018.
Portola plans to use basically the same commercial strategy and infrastructure for AndexXa as for betrixaban. Both drugs target the same audience in the hospital setting so that one sales force can sell both products. They will implement a strategy to manage the limited initial supply of AndexXa by focusing on high risk patients with intracranial bleeding or gastrointestinal bleeding that results in hemodynamic compromise; these patients have the highest mortality rates. Once they have an adequate supply from the Generation 2 manufacturing process, they expect to expand hospital targeting in the US.
Financial Issues
Portola has been well financed and this has allowed it to keep almost all the commercial rights to its two drugs. It has licensed betrixaban in Japan, but has kept all other worldwide commercial rights. As of December 31, 2016 Portola had cash and equivalents of $319 million. On February 2, 2017 a royalty agreement on AndexXa with Health Care Partners brought in $50 million upfront which gives Portola $369 million of cash resources for 2017. Portola does not give specific guidance on cash burn, but indicates that manufacturing (which is included in R&D) will be the main driver. In 2016, Portola reported that in 2016 R&D expenses were $249 million and S, G & A was $58 million for total operating expenses of $307 million. Operating cash burn in 2016 was $196 million.
The Health Care Partners deal will provide another $150 million if AndexXa is approved this year or early next year. Management says that it has a cash runway into 2018, but doesn’t get more specific. With potential launch preparations and expenses, the operating burn rate could accelerate sharply in 2017 and 2018 from the $196 million in 2016. I think that Portola needs to bring in a significant amount of cash prior to mid-2018 even if this $150 million is received; it could prove reckless to wait until early to mid-2018. I would not be surprised to see Portola move to bring in $300 million or so in 2H, 2017 following the anticipated betrixaban approval and possibly the AndexXa approval. One or both approvals should cause a significant boost to the stock price.
If Portola were to raise $300 million in an equity offering, at the current price of $40, it would increase the share count by 7.5 million to 64.0 million. Management also has the option to partner or co-promote betrixaban and andexamet in almost all regions of the world. So far, it has only licensed rights to betrixaban in Japan. Raising cash should be relatively easy if approval is gained for betrixaban and andexamet. Slippage for unanticipated reasons could result in stock pressure, a cash squeeze and the need to finance at lower prices. This worries me, but I do think that betrixaban and andexamet both will be approved in the US and the EU by early 2018.
The Stock Has Nearly Doubled This Year
The stock has experienced wild swings in prices since it came public on May 20, 2013. It has nearly doubled so far this year. However, it is below the $56.90 price that it sold at on September 14, 2015 when it was expected that both betrixaban and andexamet would be approved in 2H, 2016. The bottom fell out of the stock when timing of or actual approval of betrixaban and andexamet came under question so that on October 31, 2016, the stock hit a low of $17.65. We are now looking at essentially the same fundamental outlook as on September 14, 2015 when the stock reached $56.90 and yet the stock is selling at a lesser price of $40.05. There is sometimes a reluctance to consider purchasing a stock after a strong move such as we have seen in 2017, but this discussion argues against that. It suggests that the move is basically just a recovery from an abnormally depressed valuation.

Setting a Price Target
It is always difficult to set a price target for a pre-commercial biotechnology company because of the enormous uncertainties relating to clinical trial outcomes, regulatory action and then commercialization. There are a huge number of unconstrained variables. The most common valuation approach used by Wall Street is to construct a P&L for five years or so and then use a discounted cash flow model to determine a net present value for the stock. This looks impressive, but I think that because of the huge number of variables that it is virtually worthless. Oftentimes, analysts determine the price target that they want and then do a calculation that gives them that number.
I have great humility in coming up with a price target, but I feel compelled to make an effort. My approach is to focus on sales at some period of time, usually five years out. While sales are difficult to project, they have much higher predictability than a model based on discounted cash flow. Choosing a five year time frame also gets past focusing on the first year or two of launch when reimbursement issues can distort sales (usually on the downside). Later in this report, I detail how I come up with the sales projections shown in the following table that projects 2022 sales for Portola of $2.235 billion.

Having come up with a sales estimate, the next question is how to translate that into a stock price. The first step is to look at the market capitalization of a range of other biotechnology companies that are in the commercial stage and then calculate the ratio of market capitalization to sales. The following table shows the market capitalization for a group of 13 commercial stage companies ranging from mature companies lie Amgen and Biogen to earlier stage companies. For each, I calculate the ratio of current market capitalization to 2017 projected sales.
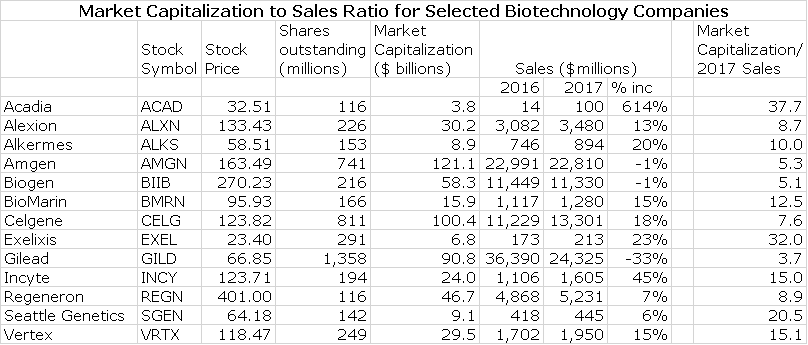
The above table show that the most mature biotechnology companies, Amgen and Biogen, sell at about 5 times projected 2017 sales. The large but less mature companies like Alexion, Celgene and Regeneron sell at 8 to 9 times. BioMarin and Incyte are viewed as still relatively early stage companies and the market values them at 13 to 15 times projected 2017 sales.
I next assume that the myriad of macro-economic and micro-economic issues that affect biotechnology stocks will be the same in 2021 as they are now. This is highly unlikely, but I don’t know what other assumption can be made. I think that in 2021, that investors will be more inclined to look at Portola as they now look at BioMarin and InCyte and if so, this would lead to a market capitalization of 13 to 15 times projected sales and a Portola market capitalization of $29 to $34 billion. For subsequent discussion, I will use the mid-point of this range which $31.5 million.
In order to calculate the stock price target for 2021, we next have to estimate the number of shares that will be outstanding. I previously indicated that I thought that Portola might raise $300 million later this year by issuing 7.5 million shares that could bring the share count to 64.0 million shares. There will likely be the need for additional capital as Portola continues a global launch of its two products. I am arbitrarily assuming that this will result in the issuance of another 10.0 million shares over time so that the share count in 2021 is 74.0 million shares. Dividing the estimated market capitalization of $31.5 billion by 74.0 million shares results in a 2021 price target of $426 per share.
I want to issue a note of caution that while these calculations give the impression of great precision, they are anything but. You should take these calculations only as an indicator of trend and magnitude. I would be the most amazed man on Planet Earth if my sales projections and price target projections are even remotely close. That said, I want to leave you with the thought that there is the potential for explosive upside in this stock if betrixaban and andexamet are approved commercialized.
Risk Factors
It is obviously not a slam dunk that the FDA will approve betrixaban at its June 24, 2017 PDUFA date. There are some knowledgeable investors who feel that the FDA will require the company to do a new phase 3 trial. I think this could cause a dramatic drop in the stock price.
It is impossible for an outside investor to assess the ease with which Portola can address the manufacturing issues in the andexamet CRL. Sometimes these issues can drag on for much longer than a company expects. The combination of the requirement for a new phase 3 trial for betrixaban and a further delay in AndexXa approval could potentially drive the stock to the $15 to $20 range.
The final risk factor I would cite is that Johnson & Johnson has decided to do another phase 3 trial with Xarelto in acute medically ill patients. MAGELLAN, the first clinical trial in this indication, reached the primary efficacy endpoint, but was rejected because of excessive bleeding risk. In 2014, JNJ began the MARINER trial which is designed much like APEX. It plans to enroll 12,000 patients and CLinTrials.gov indicated that the completion of the trial is scheduled for May 9, 2018. If this timetable is met, the NDA could be filed in 2H, 2018 and assuming a positive outcome, approval could come in mid-2019.
I think that the available data suggests that betrixaban is safer than Xarelto and it is a true once a day drug whereas Xarelto is not. However, even if betrixaban is a better drug, JNJ would have a major marketing advantage in that the existing huge sales base of Xarelto allows JNJ to offer huge rebates. Approval of Xarelto in mid- 2019 2019 could cause the sales curve of betrixaban to flatten out in 2020 and make my sales projections for subsequent years too high.
JNJ must believe that the design of the MARINER trial will produce positive results. However, at this time, I am not figuring such success into my sales projections primarily because of the results of the MAGELLAN trial provide the only data insight into Xarelto and that trial failed. It is hard for me to presume that the new trial will succeed while MAGELLAN failed and how results might measure up to data from betrixaban’s APEX trial. Success in the MARINER trial would likely cause me to lower my price target for betrixaban, but even so the stock has enormous upside potential from current levels.
Basic Biology Background on Coagulation
Background on Coagulation and Thrombosis (Blood Clots)
The blood clotting process (hemostasis) is essential to survival, but excessive clotting can lead to life threatening situations. When this occurs, it is necessary to give drugs which reduce the ability of blood to clot. These drugs are referred to as anticoagulants or anti-thrombotics.
A blood clot is an aggregation of platelets within a blood vessel that is usually caused by: (1) slowing of blood flow, (2) damage to a blood vessel wall that causes the generation of clotting factors, or (3) hypercoagulability arising from genetic disorders. Abnormal blood clot formation can block or partially block blood flow to tissues (ischemia) and this ischemia can cause tissue damage (infarction).
The most frequent medical problems caused by blood clots are:
- The formation of blood clots in the arteries of the heart can cause heart attacks.
- Blood clots that break off from vessel walls (usually veins in the legs), travel through the circulatory system and lodge in smaller vessels of the body; these are called thromboemboli. If they lodge in the brain they can cause strokes and if they lodge in the lungs they can cause pulmonary embolisms which impedes the flow of oxygenated blood.
- Another major clot related problem stems from atrial fibrillation. This is an abnormal rhythm in the atrial chamber of the heart which causes a fluttering effect that results in a slowing of the blood flow. This can cause the formation of blood clots that then travel through the circulatory system into the brain and lungs.
The reasons for clots forming in the arterial system and the venous system are usually different. Arterial clots are often associated with plaque formation; the buildup of cholesterol and other lipids in the vessel wall that can rupture, cause clots to form and lead to ischemic disease such as acute coronary syndrome, stroke and peripheral artery disease. Cardiovascular procedures such as stenting can also damage vessel walls and lead to the formation of clots.
This report focuses on drugs used to:
- Prevent or treat clots in the veins. These can result from the slowing of blood flow such as can occur in immobilized patients following surgery or hospitalization for other diseases, and
- Blood clots caused by atrial fibrillation which cause stasis or slowing of blood flow in the atrial chamber of the heart.
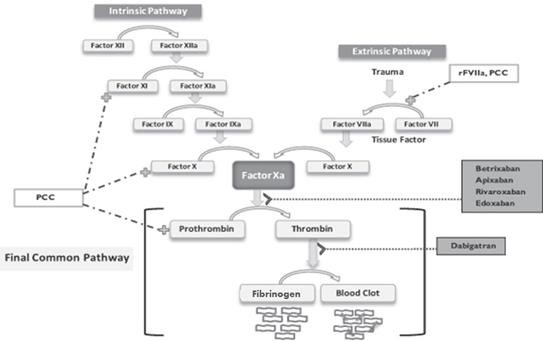
The Clotting Cascade Leads to Clot Formation
Anticoagulant drugs interact with proteins that interact with each other in a complicated clotting cascade that leads to formation of a clot. The essential cells in the clotting process are platelets which circulate in the blood. Platelets must be activated and aggregate at the site at which a clot is formed. Molecules on the surface of platelets then work in coordination with a second system that involves the actions of multiple proteins that are continually circulating in the blood called clotting factors which work together in a process that is known as the clotting cascade. As you will see shortly, this is an incredibly complex system. I will try to convey the basics of this system, but it is one in which I have only rudimentary understanding.
The essential function of the clotting cascade is to form fibrin, which wraps a mesh between the aggregated platelets to stabilize the clot. The clotting cascade involves numerous clotting factors, each of which has an active and inactive form. (The letter 'a' is used to denote an active form of a factor.) Following activation, a factor will then activate the next factor in a lengthy sequence until fibrin is formed. There are multiple interactions in the cascade that are required for coagulation to work properly.
The coagulation cascade takes place at the site where platelets have aggregated. The key steps are as follows:
- Tissue factor and factor VIIa activate factor X, forming factor Xa (remember the small a means it is activated).
- Factor Xa is then able to activate factor II (also known as prothrombin) to form factor IIa (thrombin).
- Thrombin converts factor I (fibrinogen) to factor Ia (fibrin). Fibrin forms a mesh with the platelets that plugs the break in the vessel wall.
- The fibrin mesh is further stabilized by factor XIII, which sews up the clot (much like forming an intricate network of cross-stitched strands of fibrin).
- Factor V and factor VIII accelerate the conversion of factor X to factor Xa by factor IXa (this is done by factor VIII) and accelerate the conversion of prothrombin to thrombin as done by factor Xa.
This dizzying sequence is shown below:
Older Drugs Used for Treating Clots
Prior to 2010 when a number of important new drugs began to come to market, several older drugs were the backbone of therapy to treat or prevent clots. They were:
- heparin (unfractionated),
- the fractionated low molecular weight heparins (LMWH)
- warfarin which is generally referred to by the brand name Coumadin
- aspirin and Plavix (clopidrogel)
Heparin is a naturally-occurring anticoagulant produced by mammalian cells that is harvested from the intestines of pigs. It acts by preventing the formation of clots. It does not work directly to break down clots that have already formed; instead, it allows the body's natural clot dissolving mechanisms to work normally to break down clots that have formed. Heparin needs to be given by continual infusion and this means that it is generally used for short term anticoagulation in the hospital setting for treating acute coronary syndrome, atrial fibrillation, deep vein thrombosis and pulmonary embolism, cardiopulmonary bypass for heart surgery, and extracorporeal life support and hemofiltration. It requires careful monitoring to make sure that correct amounts are given.
The low molecular weight heparins (LMWH) are comprised of smaller (fractionated) sub-segments of the heparin molecule. Some of the better known LWMH products are Lovenox (enoxaparin) and Fragmin (dalteparin). They can be given subcutaneously and unlike heparin do not require continual monitoring. Because LMWH drugs have more predictable pharmacokinetic and anticoagulant effects, LMWH is recommended over unfractionated heparin for patients with massive pulmonary embolism and for initial treatment of deep vein thrombosis.
Prophylactic treatment of hospitalized medical patients with LMWH results in a significant reduction of risk for symptomatic deep vein thrombosis. Because it can be given subcutaneously and does not require monitoring, LMWH can be used as outpatient treatment in conditions such as deep vein thrombosis or pulmonary embolism unlike heparin which can only be given in a hospital. However, the need to give one or two subcutaneous injections per day limits effective utilization in the outpatient setting.
Warfarin or Coumadin (the brand name of the most widely used warfarin product) was long the standard of care in treating clots that form from atrial fibrillation. Frequently, patients requiring long term anticoagulation are started on LMWH and then switched to oral warfarin. Warfarin inhibits the synthesis of clotting factors in the clotting cascade that are biologically dependent on vitamin K. It is a difficult drug to use. It requires tight monitoring to titrate to the correct dosage and even after the right dosage is determined, the patient must have blood drawn every two weeks to make sure that the dosage remains correct. It has frequent and troublesome interactions with food and other drugs. Like LMWH it has a slow onset of action of a day or so.
Aspirin and Plavix (clopidrogel) are oral agents that are the mainstays of managing arterial clots. They are used prophylactically to reduce the risk of heart attacks. These drugs works on different biological targets: aspirin on the COX-1 enzyme and Plavix on the P2Y12 receptor on platelets. Both mechanisms prevent the aggregation of platelets that is necessary to form blood clots.
Newer Anticoagulants
The older anticoagulants played a critical role in cardiovascular medicine saving countless lives each year and were reasonably safe. However, there remained an unmet need for new products that could be given orally to extend treatment for which the injectable formulations of heparin and LMWH are not practical. There was also a need for new products as effective as warfarin, that didn’t have all of the dosage and monitoring issues of this drug. This opened the door to newer drugs like Pradaxa (dabigatran), Xarelto (rivaroxaban), Eliquis (apixaban) and Savaysa (edoxaban). Pradaxa is a direct thrombin inhibitor. Xarelto, Eliquis and Savaysa are factor Xa inhibitors that work in the clotting cascade upstream of thrombin, which needs factor Xa for its formation. Both approaches reduce the amount of thrombin formed and consequently the production of fibrin.
The time of US approvals for preventing strokes due to atrial fibrillation for these three drugs were as follows:
- Pradaxa-October 19. 2010
- Xarelto-November 4, 2011
- Eliquis-December 28, 2012
- Savaysa-January 8, 2015
The time of US approvals for treating deep vein thrombosis and pulmonary embolism were as follows:
- Xarelto-November 2, 2012
- Pradaxa-April 7, 2014
- Eliquis-August 21, 2014
Other approved indications were:
- Eliquis- for reducing the risk of clots following hip or knee surgery March 18, 2014
- Pradaxa-Prophylaxis of deep vein thrombosis and pulmonary embolism after hip replacement surgery November 23, 2015
One of the major advantages offered by these new drugs was that they were just more convenient to use. Drugs are only effective if patients use them correctly and this was the problem with older drugs. The need to infuse heparin limits its use to the acute hospital setting. While low molecular weight heparins can be used outside the hospital, one or two injections per day is a difficult regimen for patients to adhere to. Coumadin (warfarin) is just a royal pain in the neck to initially titrate and then maintain at the right dosage. Pradaxa, Xarelto, Eliquis and Savaysa are once or twice a day oral drugs that are as effective as low molecular weight heparin or warfarin but require no significant titration, no monitoring, have few food and drug interactions and have a fast onset of action.
A December 23, 2010 editorial in the New England Journal of Medicine had this to say about these new drugs.
“The oral factor Xa inhibitors (Xarelto and Eliquis) represent a major advance in the prevention and treatment of thromboembolic disease. …… The potential impact of these oral, highly specific, fixed-dose drugs that do not require routine monitoring will no doubt be substantial. Currently, millions of people worldwide are relegated to receiving no therapy or therapy that has been proven to be ineffective, because they lack access to the monitoring expertise needed to safely and effectively administer warfarin. It is conceivable that the oral factor Xa inhibitors, as compared with warfarin, will prove to be safer in clinical practice because they are administered in fixed doses, do not interfere with diet, and have fewer interactions with other drugs.
Alternatives to warfarin have been long awaited. The oral factor Xa inhibitors show great promise. The reversibility of the drugs' effects and the ability to measure the anticoagulant effect in specific situations will continue to be highly desirable features and will help to allay physicians' concerns. If these novel, breakthrough, oral anticoagulant drugs prove to be effective across the broad spectrum of patients in routine care and are conscientiously priced, the worldwide impact will be huge.”
What is the Role of Betrixaban?
Fulfills Unmet Medical Need
Betrixaban is an oral factor Xa inhibitor like Xarelto, Eliquis and Savaysa. Very importantly, it is targeting an important segment of the anticoagulant market for which Pradaxa, Xarelto, Eliquis and Savaysa haven’t been approved. Portola has positioned betrixaban to be the first anticoagulant drug approved for extended duration, venous thromboembolism (VTE) prophylaxis in acute medically ill patients. These are patients who are hospitalized for non-surgical conditions, such as heart failure, stroke, infection, rheumatic disorders and pulmonary disorders. Because of restricted mobility and other risk factors, they have an increased risk for VTE, both in the hospital and after discharge.
Current Treatment of Acute Medically Ill Patients
In the acute medically ill setting in the hospital, heparin and LMWH are the drugs that are currently used. They have shown in clinical trials that they can reduce the rate of venous and pulmonary thromboembolism by more than 50% without major bleeding in high risk patients who are characterized by
- Several days of restricted mobility,
- age,
- an elevated blood marker known as D-dimer,
- previous VTE event,
- family history of VTE,
- smoking,
- hormonal therapy and others.
These patients are typically treated for 6 to 14 days, most of which is in the hospital. Almost all hospitalized non-surgical patients have at least one of these risk factors, and approximately two-thirds have two or more.
The important therapeutic niche for betrixaban is to continue treatment for an extended period after the patient is discharged from the hospital. Heparin is infused so that it can only be used in the hospital and subcutaneous, twice a day administration of LMWH is a substantial barrier to use after discharge. They just aren’t practical in the out-patient setting. There are no other drugs approved in this setting and this represents a significant unmet medical need.
There is a material risk of VTE after discharge and no oral drugs have been approved for this indication. Johnson & Johnson conducted a large trial in acute medically ill patients that was unsuccessful, but provided important data. The MAGELLAN study of 8,101 patients showed that the rate of VTE-related death for the 10-day period while the patients were in the hospital receiving anticoagulation therapy was 0.2%. Importantly In the 25 day period after discharge, the VTE related death rate for patients who did not receive anticoagulation treatment, was four times higher at 0.8%.This highlights the need for extended duration of anticoagulation after discharge.
Why Aren’t Xarelto and Eliquis Used in this Setting?
There have been three previous large trials involving three different drugs. The drugs and trials were as follows:
- Lovenox- the Extended Clinical Prophylaxis in Acutely Ill Medical Patients (EXCLAIM) trial of 4,726 patients
- Eliquis- the Apixaban Dosing to Optimize Protection from Thrombosis (ADOPT) trial of 6,524 patients and
- Xarelto-the Multicenter, Randomized, Parallel Group Efficacy and Safety Study for the Prevention of Venous Thromboembolism in Hospitalized Acutely Ill Medical Patients Comparing Rivaroxaban with Enoxaparin (MAGELLAN) trial of 8,211 patients
The MAGELLAN and EXCLAIM trials both met the primary efficacy endpoint of decreased VTE in acute medically ill patients. However, the bleeding risk was not acceptable so approval was denied based on a benefit to risk ratio. The ADOPT trial of Eliquis did not demonstrate significant clinical efficacy, but the incidence of major bleeding was lower than was seen in MAGELLAN.
How Might Betrixaban Succeed Where Eliquis and Xarelto have Failed?
Betrixaban, Xarelto and Eliquis all have the same mechanism of action, they inhibit factor Xa. The obvious question is why betrixaban might succeed where Eliquis and Xarelto have failed. Phase 3 clinical trials like MAGELLAN and ADOPT can fail because of drug properties or the dosing of the drug (not all factor Xa inhibitors are the same). There are intrinsic properties of betrixaban that may make it more effective than Xarelto and Eliquis in this setting. Importantly drug trials can also fail because of patient selection for the trial and trial design. Even though all three drugs work through factor Xa inhibition, these issues resulted in betrixaban succeeding where the other two drugs failed.
Design of the Phase 3 APEX Trial of Betrixaban in Acute Medically Ill Patients
Portola used its detailed knowledge of betrixaban’ s drug properties and phase 2 clinical experience and took into consideration learnings from the MAGELLAN and ADOPT trials to design the phase 3 APEX trial. It was designed to demonstrate the safety and efficacy of betrixaban for extended duration, VTE prophylaxis starting with a hospital stay and continuing for a 35 day period following discharge. The patients were acute medically ill patients with prospectively defined risk factors. It was a randomized trial comparing:
- a once-daily dose of 80 mg of betrixaban for 35 days (starting in the hospital and continuing after discharge)
- to in-hospital administration of 40 mg of enoxaparin once daily for 6 to 14 days followed by placebo for the remainder of the study period.
Between 35 and 42 days, ultrasound imaging was used to detect new clots. The primary endpoint was to show superiority of betrixaban versus enoxaparin as determined by the reduction of VTE related events at 35 days plus. This design is shown in the following schematic.
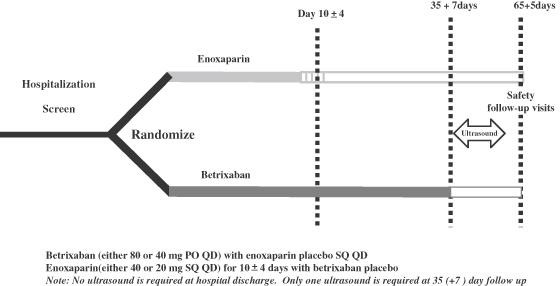
From March 2012 through November 2015, 8,589 patients were screened at 460 sites in 35 countries. Of these patients, 7,513 were found to be eligible to participate in the study and underwent randomization (3,759 in the betrixaban group and 3,754 in the enoxaparin group). Protocol changes were made in June 2014 to target (enrich) patients at high risk for VTE. This required the presence of an elevated d-dimer level or an age of at least 75 years as an entry criterion. The statistical plan was modified to create two cohorts.
- Cohort 1 included 3,870 patients who had an elevated baseline d-dimer level (two times or more the upper limit of the normal range),
- Cohort 2 included the patients in cohort 1 plus those who were 75 years of age or older for a total of 5,735 patients
- Cohort 3 was all patients enrolled in the trial including those enrolled before June 2014 for an overall total of 6,286 patients.
The primary efficacy outcome was a composite of:
- Either asymptomatic or symptomatic deep-vein thrombosis between day 32 and day 47,
- Symptomatic nonfatal pulmonary embolism, or death from venous thromboembolism between day 1 and day 42.
The principal safety outcome was the occurrence of major bleeding at any point until 7 days after the discontinuation of all study medications.
The APEX Trial Appeared to Fail, But………
The three cohorts that were included in the efficacy analysis were pre-specified in a procedure with a fixed hierarchical sequence to adjust for the type I error rate. If betrixaban was superior to enoxaparin on the primary endpoint in cohort 1, the primary efficacy endpoint would then be tested in cohort 2 and if superiority was shown then the primary efficacy endpoint would be tested in the overall study population which was cohort 3. The statistical design specified that if superiority was not met in cohort 1 then analyses of data in the other two cohorts would be only exploratory. This statistical design produced a problem.
The decision to perform hierarchical testing of trial outcomes in subgroups of the overall trial population was based on the expectation that patients with an elevated d-dimer level or an age of at least 75 years would represent a subgroup enriched for both a greater risk of venous thromboembolism and it was expected that they would have a greater benefit from extended-duration antithrombotic therapy. These expectations were based on data for similar patients who were enrolled in the MAGELLAN trial of rivaroxaban.
The primary efficacy outcomes for each cohort were as follows:
- In cohort 1, the primary efficacy outcome occurred in 6.9% of the betrixaban group and 8.5% of the enoxaparin group. The relative risk in the betrixaban group was 0.81 (95% confidence interval [CI], 0.65 to 1.00; P=0.054). This first test in the sequence of cohorts did not meet the pre-specified threshold for statistical significance; therefore, all subsequent pre-specified efficacy outcomes were considered to be exploratory and were not used to draw conclusions regarding statistical significance.
- In cohort 2, the primary efficacy outcome occurred in 5.6% of the betrixaban group and 7.1% of the enoxaparin group (relative risk, 0.80; 95% CI, 0.66 to 0.98; P=0.03).
- In cohort 3 (the overall population), the primary efficacy outcome occurred in 5.3% and 7.0% of the patients, respectively (relative risk, 0.76; 95% CI, 0.63 to 0.92; P=0.006).
Safety results relating to bleeding were very positive. In the overall safety population, major bleeding occurred in 0.7% of the betrixaban group and 0.6% of the enoxaparin group (relative risk, 1.19; 95% CI, 0.67 to 2.12; P=0.55). There were two cases of intracranial bleeding in the betrixaban group and seven in the enoxaparin group. There was one case of fatal bleeding in each group. Unlike enoxaparin in the EXCLAIM trial, apixaban in the ADOPT trial, and rivaroxaban in the MAGELLAN trial, the use of extended-duration betrixaban in APEX was not associated with significantly more major bleeding than standard-duration enoxaparin.
Portola Soldiers On; Upon More Detailed Analysis the Trial Could Be Judged a Success
Based on the trial design, the APEX trial failed as the p value in cohort 1 was 0.054. However, there was a dispute as to whether one patient in the control group had experienced an event. The CRO conducting the trial did not judge that an event had occurred, but this was disputed by Portola and some KOLS. If the patient had been judged to have had an event, the trial would have been successful the primary endpoint would have been reached with p=0.048.
The results in cohort 1 are perplexing as it was expected that sicker patients would benefit more. Cohort 2 included the 3,870 patients from cohort 1 and another 1,875 patients who because of age were thought to be at higher risk. It was unexpected that this group achieved statistical significance on the primary endpoint of p=0.03 when cohort 1 did not. Cohort 3 included cohort 2 and 551other less sick patients and in this large patient group the p value was a very impressive 0.006. Surprisingly, patients who prospectively were thought to be less sick patients appeared to do better. In addition to the narrow miss in cohort 1, the secondary endpoints generally supported superior efficacy over enoxaparin. Very importantly, the safety data showed no risk of major bleeding versus enoxaparin and even hinted at slightly less major bleed issues with betrixaban.
What Will FDA Do?
Many investors felt that even with this narrow miss on the primary efficacy endpoint and encouraging safety data on major bleeds that the FDA would remain committed to statistical purity and ask for an additional confirmatory phase 3 trial. However, this view appears to have been incorrect. Portola soldiered on and submitted an NDA on October 15, 2016. On December 23, 2016 the FDA accepted this filing and designated betrixaban for priority review which signals that the FDA believes there is an unmet medical need and promises to review the NDA in six months rather than the standard ten. This established a PDUFA date of June 24, 2017.
On February 8, 2017, Portola announced that the FDA informed the company that it had not identified any issues that require public discussion at an FDA Advisory Committee. Therefore FDA does not plan to hold an Advisory Committee to discuss betrixaban NDA, before the June 24, 2017 PDUFA date. The Company and most investors interpreted this as a positive.
The time may be right for an application like betrixaban, where the primary endpoint failed to meet the p<.05 threshold but came oh so close to be the basis for approval. The primary efficacy point that showed a p value of 0.006 for all patients in the trial strongly suggests that the drug is quite effective. Also the pre-specified subgroup analyses were positive as was the safety data on the key issue of major bleeding. In the era of Trump, there is also pressure for the FDA may to be more flexible and less statistically pure. I have strong confidence that betrixaban will be approved on June 24, 2017 but obviously this is not a sure thing.
Sales Projections for Betrixaban
There are estimated to be about 11 million acute care patients in the US at risk annually for a VTE. According to Portola, about 60% of those patients meet the enrollment criteria for the APEX study. This is about 6.6 million patients. Of these only 10% or so receive a short term course of seven days of prophylaxis with heparin or enoxaparin. I estimate that betrixaban could penetrate 60% of this 6.6 million patient target market within five years. This would result in 4.0 million patient treatments per year.
I am further assuming that betrixaban will be prescribed for 35 days so that there will be 140 million patient days of therapy per year. The current list price of the factor Xa inhibitors is about $9.00 per day but after estimated discounts of 30%, it is probably closer to $6.30. I arrive at my five year sales estimate of $925 million by multiplying 144 million patient days by $6.30.
The usual rule of thumb is that the addressable patient population in the rest of the world, principally in the EU, is about the same as the US. However, pricing is much tighter internationally so that the price of therapy per day is about 50% of the US or $3.15 per day. This results in an ROW sales forecast of about $400 million.
Addressing the Medical Need for Antidotes to Factor Xa Inhibitors and other Anticoagulant Drugs
Overview
The entry of factor Xa inhibitor drugs has led to broad usage as they reduced the monitoring and dosing issues associate with warfarin and the dosing issues with LMWH. While they were an important advance in anticoagulation therapy, they presented a major drawback in that there was no antidote to reduce major bleeding episodes that occur when patients are on the drugs. Hence, many physicians and patients still perceive warfarin as safer because there are reversal strategies for when major bleeding occurs. The need for an antidote for the factor Xa inhibitor drugs is critical. Portola has developed AndexXa (andexamet) as a way to reverse the effects of the factor Xa drugs. AndexXa has a pronounced first mover advantage.
In October 2015, idarucizumab, a humanized monoclonal antibody antigen-binding fragment that binds to dabigatran, received expedited approval from the Food and Drug Administration for use in the United States. This drug is not an effective antidote for the factor Xa inhibitors.
The privately owned company, Perosphere is developing a drug called ciraparantag that is claimed to be a universal antidote for LMWH drugs, the thrombin inhibitor Pradaxa and the factor Xa inhibitors. There is not a great deal of clinical information on the drug. According to the company website, it appears to be in phase 2 trials for reversing LMWH and in pre-clinical trials for reversing Pradaxa, Xarelto and Eliquis. It could be four years or more behind andexamet. The FDA granted ciraparantag fast track approval status in 2015.
Design of the Andexamet Molecule
Portola used recombinant genetic technology to skillfully design andexamet, a protein that is structurally similar to naturally occurring factor Xa. Like factor Xa, it binds tightly to factor Xa inhibitors such as Xarelto, Eliquis, Savaysa and betrixaban as well as non-direct factor Xa inhibitors such as the LMWH drugs enoxaparin and fondaparinux. Andexamet is a decoy molecule which by binding to these drugs prevents them from binding to factor Xa. Thus the amount of drug in the blood available to produce an anti-coagulant effect is sharply reduced.
Portola made modifications to naturally occurring factor Xa so that andexamet:
- Unlike factor Xa, it does not convert prothrombin to thrombin so that it has no effect on coagulation.
- Like factor Xa, it can bind tightly to the factor Xa inhibitors-Xarelto, Eliquis and edoxaban well as non-direct factor Xa inhibitors such as enoxaparin and fondaparinux.
- Another adjustment was made so that it has no effect on the binding and coagulation effect of naturally occurring factor Xa.
- And still another adjustment was made so that it would not be recognized by the body as foreign and trigger an immune reaction.
Design of the Phase 3 Program
Following encouraging phase 2 trials, Portola held an end of phase 2 meeting to consult with the FDA on the design of a phase 3 program. Because FDA had designated andexamet as a breakthrough therapy the interaction with the agency was extensive. A major issue to be resolved was whether andexamet could be approved on the basis of using surrogate, pharmacodynamic markers as the primary efficacy endpoint. A randomized controlled trial would present ethical challenges. Patients presenting with major bleeding are in a life threatening situation in which the use of an experimental drug might worsen the bleeding or cause dangerous side effects. .
The FDA gave the nod to a phase 3 design in which Portola conducted two randomized, double-blind, placebo-controlled studies. These studies were done in healthy volunteers. The participants were older than those in typical study populations, with an age range of 50 to 75 years to more closely approximate the age of the population at risk One trial (ANNEXA-A) was to reverse apixaban and the second (ANNEXA-R) was to reverse rivaroxaban. Volunteers were initially given the standard dose of apixaban or rivaroxaban for four days to achieve steady-state concentrations of the drug in the blood. This was followed by treatment with andexamet to see if it could then turn off the effect of these drugs.
The goal was to rapidly and significantly reduce the anti–factor Xa activity of these drugs, This was determined in a test that measured unbound levels of apixaban and rivaroxaban in the blood within 2 to 5 minutes after intravenous administration. This bolus infusion resulted in a rapid decrease in unbound drug concentrations in the blood for both apixaban and rivaroxaban and with that significantly decreased anti–factor Xa activity. However, an increase in both measures could be detected within 15 minutes after completion of the bolus. This was addressed by giving a subsequent two hour infusion that achieved sustained suppression.
The doses of andexanet differed between the ANNEXA-A and ANNEXA-R studies, with participants in the rivaroxaban study given twice the dose that was given in the apixaban study; these differences were based on pharmacokinetic and pharmacodynamic models that incorporated preclinical data from animal models and phase 2 dose ranging studies.
Among the apixaban-treated participants in ANNEXA-A:
- anti–factor Xa activity was reduced by 94% among those who received an andexanet bolus (24 participants), as compared with 21% among those who received placebo (9 participants) p<0.001),
- unbound apixaban concentration was reduced by 80% from 9.3 ng per milliliter to 1.9 ng per milliliter (p<0.001);
- Thrombin generation was fully restored in 100% of andexanet patients versus 11% of the participants (p<0.001) within 2 to 5 minutes.
Among the rivaroxaban-treated participants in ANNEXA-R:
- anti–factor Xa activity was reduced by 92% among those who received an andexanet bolus (27 participants), as compared with 18% among those who received placebo (14 participants) (p<0.001),
- unbound rivaroxaban concentration was reduced by82% from 23.4 ng per milliliter to 4.2 ng per milliliter (9<0.001);
- Thrombin generation was fully restored in 96% versus 7% of the participants (p<0.001).
There were no serious adverse events and no thrombotic complications in the 101 participants who received andexanet. Non-neutralizing antibodies were noted in 17% of the participants, but did not appear to be clinically significant.
The strong p values on the surrogate markers and the positive safety data led to the filing on the BLA on December 28, 2015. The FDA accepted the filing on February 17, 2016 and set a PDUFA data of August 7, 2016. Unfortunately, the FDA issued a Complete Response Letter on August 7, 2016 which Portola is now addressing (more of this later).
Design of the Phase 4 ANNEXA-4 Study
As a condition of approval, Portola agreed to do a post approval, phase 4 study in patients receiving either apixaban, rivaroxaban, edoxaban or enoxaparin and who then experienced acute major bleeding that was potentially life-threatening. The ANNEXA-A and ANNEXA-R phase 3 studies were conducted in healthy volunteers. The FDA wanted to see data involving patients experiencing major bleeds in a real life setting. The phase 4 ANNEXA-4 (Andexanet Alfa, a Novel Antidote to the Anticoagulation Effects of FXA Inhibitors) study began on April 10, 2015 with patients enrolling at 20 centers in the United States, 1 center in the United Kingdom, and 1 center in Canada For ethical reasons, the study was not randomized.
The study was designed to use the same dosing schedule as that used in ANNEXA-A and ANNEXA-R. All patients initially received andexanet alfa as a bolus dose of andexanet alfa over 30 minutes followed by a two-hour infusion. One of the complications of administration is that the dose chosen differs based on the anticoagulant the patient is taking and the time from the last administration. This raises concern as to whether this information can be accurately determined in a busy emergency room, where the patient is likely to present and could result in under or overdosing.
To date, ANNEXA-4 has enrolled more than 170 patients of the approximately 350 patients targeted for inclusion. Patients were evaluated for 30 days following andexanet administration. The co-primary efficacy endpoints were the percent change in anti-factor Xa activity at two hours and assessment of hemostasis (stopping of bleeding) over 12 hours following the infusion. Hemostatic efficacy was assessed by an independent adjudication committee as either excellent, good or poor/none.
Interim Results for 67 Patients as Reported in the New England Journal of Medicine (NEJM)
In the September 23, 2016 edition of the NEJM interim data was presented on 67 patients for whom data were complete as of June 17, 2016. Of the 67 patients, 32 were receiving rivaroxaban, 31 were receiving apixaban and 4 were receiving enoxaparin. The primary site of major bleeding in these patients was gastrointestinal in 33 patients (49%) and intracranial in 28 (42%), with other bleeding sites in 6 (9%). The mean time from presentation in the emergency department to the initiation of the andexanet bolus was 4.8±1.9 hours.
All patients were evaluated for safety but 20 were excluded from efficacy measurements because of low levels of anti–factor Xa activity at bassline or missing data. This left 47 patients who were evaluable for efficacy. The study is ongoing and will enroll patients until there are 162 patients available for the efficacy analysis, with an expected safety population of approximately 350.
Of the 47 patients in the efficacy population, 37 were adjudicated as having excellent or good hemostasis (79%; 95% CI, 64 to 89), with 31 patients adjudicated as having excellent hemostasis and 6 as having good hemostasis, 12 hours after the andexanet infusion. One patient could not be evaluated because of an administrative issue. Of the 9 patients who were adjudicated as having poor or no hemostatic efficacy, 5 were receiving rivaroxaban and 4 apixaban. The rates of excellent or good efficacy were 84% for gastrointestinal bleeding and 80% for intracranial bleeding
Of the 67 patients in the safety population, there were no infusion reactions, no antibodies to factors Xa and no neutralizing antibodies to andexanet. Thrombotic events occurred in 12 patients (18%), including 1 with myocardial infarction, 5 with stroke, 7 with deep-vein thrombosis, and 1 with pulmonary embolism, with some patients having more than one event. Four patients had a thrombotic event within 3 days after andexanet treatment, and the rest occurred between 4 and 30 days. There were 10 deaths (15%), with 6 adjudicated as cardiovascular events and 4 as non-cardiovascular events. Anticoagulation was resumed in 18 patients (27%) within 30 days, but a therapeutic dose of anticoagulation was restarted before the event in only 1 of the 12 patients with a thrombotic event
The authors of the study offered the following conclusion: “The site of bleeding was most often gastrointestinal or intracranial; anti–factor Xa activity was considerably elevated in most patients and, as such, was likely to be a major impediment to clinical hemostasis. The administration of an andexanet bolus and infusion resulted in rapid and substantial reversal of anti–factor Xa activity. The clinical response 12 hours after the andexanet infusion, as measured by adjudicated hemostatic efficacy with the use of predetermined criteria, was excellent or good in 79% of the patients and was consistent across a variety of subgroups. After full enrollment in the ongoing study has provided adequate statistical power, further analysis should provide details regarding the relationship between the reduction in anti–factor Xa activity and clinical hemostatic outcomes.”
Andexanet was infused over a period of 2 to 2.5 hours, and there was partial return of anti–factor Xa activity toward pretreatment values at 4 to 4.5 hours after the initiation of andexanet. Clinical hemostatic efficacy, as assessed for 12 hours after the end of the infusion, was excellent or good in a high percentage of patients. This finding supports the idea that prolonged reversal of factor Xa inhibition may not be necessary to achieve a good hemostatic response.
This preliminary report indicated that andexanet rapidly reversed anti–factor Xa activity and was not associated with serious side effects. Effective hemostasis was achieved 12 hours after an infusion of andexanet in about 80%% of the patients. Thrombotic events occurred in 18% of the patients in the safety
In a letter to the editor in the NEJM in December 2016, Portola provided an update on the 30 day risk of thrombosis in ANNEXA-4. This was on 105 patients as compared to the 67 previously reported on. The 30 day risk of thrombosis was 12.4%. Of particular note, the percentage of patients who were put back on anti-coagulant therapy after their major bleeding episode was 27% in the first 67 patients and 40% in the 105. This means that of the 38 new patients reported on, 68% were put back on their anticoagulant drug. This is very encouraging data that suggests that when a major bleed occurs that andexanet alpha can be given to control the bleed and then anticoagulant therapy can be promptly resumed.
Complete Response Letter
On August 17, 2016 Portola received a complete response letter from the FDA. The letter asked for additional information on manufacturing primarily and also referred to issues regarding labeling and post-marketing commitments. It did not ask for any new clinical data in regard to use with rivaroxaban or apixaban, which are the two major commercial opportunities. The manufacturing issues relate to analytical assays used for quality control and assurance. Apparently, the FDA would like more data collected during manufacturing. PTLA has commented that it already collects the data and the assays are in place. However, some of the assays were not fully validated or made GMP compliant as PTLA thought this could be done after approval. Portola believes that these concerns can be answered in a reasonably prompt manner.
The FDA also requested that Portola conduct phase 3 trials in healthy volunteers similar to ANNEXA-A and ANNEXA-B for edoxaban and enoxaparin. Use in these products may be left off the label but could be added later by conducting the studies after approval or using data from ANNEXA-4. These will likely be used off label.
It appears that the BLA will be resubmitted in 2Q, 2017 and I am assuming a six month review so that approval is likely by yearend in the US. This would point to a US launch in 1Q, 2018.
Manufacturing Capacity
Portola will likely be capacity constrained at launch. The Gen 1 manufacturing process was used for phase 2, 3 and 4 trials and the BLA. A larger capacity Gen 2 process was originally planned to be available at launch. However, this transition appears to be more complex than originally thought so that Gen 2 supply is not likely to be available until mid-2018. Portola is likely to be asked to do a bridging study.
Addressable Market
Based on comments from Portola and other sources, I estimate that by 2022 there will be over 2.0 million patients in the US who are on a factor Xa inhibitor drugs and 1.4 million in Europe. There are three potential segments of this population to address:
- Clinical data suggests 1% to 2% will experience a spontaneous major bleed. This equates to 20,000 to 40,000 patients in the US and 14,000 to 28,000 in the EU. KOL sources suggest that 50% of these patients may be given AndexXa five years after launch.
- 3% will require emergency surgery. This equates to 6,000 patients in the US and 4,200 in Europe. KOL sources suggest that 60% of these patients may be given AndexXa five years after launch.
- 1% to 2% will have trauma other than a major bleed or emergency surgery. This equates to 20,000 to 40,000 patients in the US and 14,000 to 28,000 in the EU. KOL sources suggest that 30% of these patients may be given AndexXa five years after launch.
Using these assumptions, I estimate that there will be 19,600 to 35,600 patients treated in the US in 2022 and 14,000 to 25,000 in the EU. Let’s take the mid-point of these ranges and estimate that 28,000 patients will be treated in the US and 19,500 in the EU.
Portola has not commented on the price that it will charge for AndexXa, but we can look at comparable products to come up with an estimate.
- Kcentra (non-activated 4-factor prothrombin complex concentrate) is indicated for the urgent reversal of vitamin K antagonists in adult patients with acute major bleeding or who need for urgent surgery or other invasive procedure. It is priced at $10,000 per treatment.
- Voraxaze (glucarpidase) is used in patients who develop kidney failure while receiving high doses of methotrexate. It is an enzyme that breaks down methotrexate in the body so the drug can be easily eliminated when the kidneys are not working properly. It is priced at $50,000 per treatment.
- Praxbind (idarucizumab) which was approved in 2015 is the antidote for the anticoagulant Pradaxa (dabigatran) which inhibits thrombin rather than factor Xa. Praxbind is priced at $3,500 per treatment.
We have a range of potential prices from comparable products that fall in a range of $3,500 to $50,000 per patient. I think that I would rule out the $3,500 price for Praxbind because Boehringer Ingleheim developed both it and Pradaxa. Pradaxa was locked in a battle for market share with Xarelto and Eliquis, which at that time obviously had no antidote. The low price of Praxbind afforded a safety advantage for Pradaxa so that the low price was intended to boost market share of Pradaxa.
I think that AndexXa is likely to be priced at about $20,000 per treatment. If so, the potential sales in the US in 2022 would be $560 million and in ROW about $350 million.
Five Year Sales Projections for Betrixaban and AndexXa
Overview
I have previously estimated that in 2022, its fifth year on the market, that betrixaban will achieve US sales of $925 million and ROW sales of $400 million. Similarly, I estimate that AndexXa will achieve US sales of $560 million and ROW sales of $350 million.
In order to estimate the sales for these products in years one through five, I looked at the historical sales trajectories for Xarelto and Eliquis to see how sales in years one, two, three and four compared to year five. The results are shown below:
| Sales Trajectories for Eliquis and Xarelto | ||||||
| 2012 | 2013 | 2014 | 2015 | 2016 | 2017 | |
| $ millions | ||||||
| US | ||||||
| Eliquis | 97 | 404 | 1,023 | 1,963 | 2,450 | |
| Xarelto | 239 | 864 | 1,522 | 1,868 | 2,288 | 2,500 |
| ROW | ||||||
| Eliquis | 49 | 370 | 837 | 1,380 | 1,840 | |
| Percentage of Fifth Year Sales | ||||||
| US | ||||||
| Eliquis | 9 | 21 | 42 | 80 | 100 | |
| Xarelto | 10 | 38 | 67 | 82 | 100 | 125 |
| ROW | ||||||
| Eliquis | 3 | 20 | 45 | 75 | 100 | |
The above table indicates that:
- First years sales in the US were 9% to 10% of the level of fifth year sales,
- second year were 21% to 38%,
- third year were 42% to 67%,
- and fourth year were 80% to 82%
I used these results as a rough guide to project the sales trajectory for both betrixaban and AndexXa over the first five years in the market. The results were as follows:
Tagged as andexamet, AndexXa, betrixaban, Portola, PTLA + Categorized as Company Reports, LinkedIn
Very nice write up. Thank you.
Very thorough write up. My sense is that your sales numbers are very conservative. Betrixiban, due to its PK profile is superior to both currently marketed NOACs. I believe there will be aggressive clinical development to expand the label and ultimately take market share from both Xarelto and Eliquos. I also believe that this will occur after Portola is acquired by BP which I believe will be shortly after the Betrixiban approval. BP needs accretive revenue badly. My sense is there will be multiple suitors for PTLA, maybe one of the current 4 players in the NOAC space, or maybe a new player. We shall see. The next couple of months should be interesting. Again thanks, excellent summation.
Based on the current data, betrixaban does appear to be first in class. I think that you are correct that my projections appear low if you look at this situation only in terms of product characteristics. Unfortunately, payors have a powerful say in the use of products and are motivated only by the discounts they can obtain. This gives a great edge to the entrenched players with huge sales bases.
I think that PTLA is likely to be acquired.