



Northwest Biotherapeutics: Why I Believe there is a High Probability for Approval of DCVax-L (NWBO, Buy, $0.21)
Investment Overview
There are Good Reasons to Believe that Phase 3 Trial of DCVax-L in Newly Diagnosed Glioblastoma Multiforme Will Be Successful and Lead to Approval
The phase 3 trial of DCVax-L is nearing unblinding as discussed in my November 21, 2019 report. and investors are awaiting anxiously to see if the trial will be a success and lead to the approval of DCVax-L for the treatment of newly diagnosed glioblastoma multiforme (nGBM). The stock at $0.20 per share clearly is priced for failure as the market capitalization is less than $200 million. If the market perceived that DCVax-L had a reasonable chance for approval, analysis of peer companies suggests that the market capitalization could be well in excess of $1 billion. Of course, moving from a possible to an actual approval could trigger a move much higher than $1 billion. I will discuss possible price targets in a future report.
I think that there is a very good chance that DCVax-L will be successful in its phase 3 trial and then will be approved by regulatory agencies. Key to my argument (and a primary purpose for this report) is a comparison of the clinical data that led to the approval of Novocure’s medical device Optune for nGBM versus blinded data from the DCVax-L phase 3 trial. In its phase 3 trial, Optune added to standard of care (SOC) improved median overall survival (mOS) to 24.4 months versus 19.5 months for SOC treated patients. (Please note that this data is from time of surgery rather than time of randomization for reasons I will explain shortly. All data on mOS in this report is from time of surgery unless noted otherwise.)
Blinded data based on all patients treated in the phase 3 trial of DCVax-L achieved an mOS of 23.1 months and there are very strong reasons to believe that the mOS of DCVax-L added to SOC will be meaningfully better. The percentage of patients surviving at two and three years for Optune added to SOC was significantly better than SOC alone. The blinded results from the phase 3 trial of DCVax-L were slightly better than those for Optune. Blinded results showed that 46% of patients were alive at two years and 28% at three years, which compares to 43% and 26% for Optune. Again, I would suggest that the survival percentages of DCVax-L at these time periods should be better than the blinded data.
I will show and discuss this data in more detail in this report. My important conclusion is that the improvement in mOS over SOC for DCVax-L added to SOC will be as good or better than what was seen for Optune added to SOC in its phase 3 trial. I draw the same conclusion for percentage of patients surviving at two and three years. If so, the FDA should move expeditiously to approve the product.
Proposed New FDA Guidelines for Drug Approval are Very Favorable for DCVax-L
This report also discusses proposed new FDA guidelines that are very encouraging for the approval of DCVax-L. In its draft comment, the FDA states that in the case of aggressive diseases like nGBM that it may approve a new drug on the basis of one well controlled trial. This is huge for Northwest. Aggressive short sellers have argued that the FDA would require another trial for approval. This is obviously not the case.
The FDA also proposes that an investigational drug such as DCVax-L that is being evaluated in aggressive diseases like GBM can be statistically compared to historical results seen with placebo or active therapy in past trials. Short sellers have argued that there are not enough patients in the SOC control arm of the DCVax-L phase 3 trial to execute a meaningful statistical analysis, an argument with some merit. However, by comparing to historical SOC results from other trials, a meaningful statistical analysis can be done. This is a huge positive.
I have included a more in-depth discussion of these proposed and almost certain to be implemented guidelines at the very end of this report. Also, contained in these guidelines are very important proposals that would significantly shorten the time to expand indications of DCVax-L to other solid tumors.
Enrollment Comparisons for the DCVax-L and Optune Phase 3 Trials in nGBM
The DCVax-L trial was a multi-centered phase 3 trial that compared DCVax-L added to standard of care (SOC) versus SOC alone in newly diagnosed glioblastoma multiforme. The trial enrolled 331 patients with about 232 starting on DCVax-L plus SOC and roughly 99 starting on SOC. The trial allowed patients on SOC who saw their cancer progress to be able to cross over to DCVax-L and also allowed patients who started on DCVax-L plus SOC and saw their cancer progress to continue to receive DCVax-L. This results in the following patient groupings:
- About 232 patients started on DCVax-L added to SOC.
- Roughly 99 patients started on SOC.
- Northwest has stated that 90% or about 298 patients in the trial received DCVax-L at some point.
- This indicates that about 69 patients who started on SOC received DCVax-L when their cancer progressed.
- Roughly 30 patients in the trial received only SOC.
- Within the 232 patients who started on DCVax-L an unknown number continued therapy when their disease progressed.
In the Optune trial, 466 patients received Optune plus SOC and 229 received SOC. Because it is readily apparent if a patient was wearing the Optune device, the trial was not blinded. There was no cross over allowed in the trial.
Key Comparisons of Data from the Phase 3 Trial of Optune and Blinded Data from DCVax-L Phase 3 Trial:
- Optune plus SOC reported an mOS of 24.4 months versus 19.5 months for SOC, a 4.9 month improvement. Generally, key opinion leaders regard a 4.0 month improvement in mOS in an aggressive cancer like nGBM to be a significant advance. The FDA approved Optune on this data.
- Because the DCVax-L trial remains blinded, we do not know the results for the patients who initially received DCVax-L plus SOC or those who were initially given SOC. However, we do have blinded data that shows the mOS for all 331 patients in the trial and the survival percentages at two and three year time frames.
- Comparison of blinded data from the DCVax-L trial with the phase 3 trial that led to the approval of Optune in newly diagnosed glioblastoma multiforme (nGBM) shows comparable median overall survival (mOS) results and comparable percentages of patients alive at two and three years.
- The mOS blinded data for the DCVax-L trial was 23.1 months which compares to 24.4 months seen with Optune plus SOC.
- I have looked at the results seen in the SOC control arms of six trials which enrolled 1,608 patients on SOC. The mOS for SOC in these trials had a tight range of 19.1 to 20.2 months so this seems to pretty well define what SOC can deliver. If DCVax-L had no effect in the trial, we would have expected the blinded mOS results to be 19 to 20 months, but the blinded data showed a meaningfully higher mOS of 23.1 months.
- This strongly suggests that DCVax-L drove the meaningfully higher mOS and we can hypothesize that the mOS of DCVax-L plus SOC will be meaningfully higher than 23.1 months and as good or better than that which resulted in approval of Optune.
- The percentage of Optune plus SOC patients surviving at two years was 43% and 26% at three years. The blinded results from the phase 3 trial of DCVax-L were slightly better showing 46% of patients alive at two years and 28% at three years. For SOC, the respective percentages are 27% and 16%. Again, I would hypothesize that the survival percentages with DCVax added to SOC might be as good or better than those with Optune added to SOC.
- The mOS of blinded data of 23.1 months is a 3.1 to 4.1 months improvement over the 19 to 20 months expected with SOC. As a point of reference, the chemotherapy drug temozolomide was approved in 2005 on the basis of showing a 2.5 month improvement in SOC.
Key Definitions, Abbreviations and Assumptions
- Median overall survival (mOS)is the timepoint in a clinical trial at which half of the patients have died. This has been and remains a critical endpoint for most oncology trials although the survival tail is becoming increasingly important for immune-oncology drugs like DCVax-L.
- Standard of care (SOC) in newly diagnosed glioblastoma multiforme (nGBM) currently is surgical resection followed by courses of radiation and the chemotherapy drug temozolomide.
- The blinded results of the DCVax-L trial lump together patients who started in DCVax-L plus SOC and those started on SOC. We don’t know results for each group. The Optune trial has been unblinded so that we can see and compare results for patients treated with Optune plus SOC versus those treated with SOC.
- The results published in medical journals for mOS as shown in the Optune trial are based on the time from randomization. This is also true of SOC results seen for nGBM trials from other trials that are referenced in this report.
- DCVax-L published results are shown from the time of surgery.
- The median time from histological diagnosis to randomization in the Optune trial was reported as 3.8 months. I estimate that the median time from surgery for patients in the Optune trial was about 3.5 months. This assumes that surgery occurred one to two weeks after diagnosis. I have no concrete data to support this other than anecdotal reports.
- In order to have an “apples to apples” comparison between results seen in the phase 3 trials of DCVax-L and Optune, I have increased reported mOS in the Optune trial by 3.5 months to estimate time from surgery. For example, mOS in the Optune trial was reported as 20.9 months (from time of randomization) and my adjustment increased this to 24.4 months (estimated time from surgery). I did the same for SOC results referenced from other trials. Unless expressly noted otherwise, all mOS data in this report is from time of surgery.
- Please make sure that you understand the four previous bullet points
Blinded Data from the Phase 3 Trial of DCVax-L
Northwest Biotherapeutics reported blinded results from its phase 3 trial of DCVax-L in November 2018 at the Annual Meeting of the Society for Neuro-Oncology. This trial enrolled 331 patients of whom roughly 232 began the trial on DCVax-L plus standard of care (SOC) and roughly 99 began on SOC. The results remain blinded for this trial that began in 2007 and in which the last patient was enrolled in November of 2015. Twice the Company has reported blinded data from the trial, first in 2017 and most recently on November 19, 2018. Here is some of the data reported.

The blinded results combine the outcomes of roughly 232 patients who received DCVax-L plus SOC and about 99 who received just SOC or 331 in total. Because the mOS for all 331 patients of 23.1 months from time of surgery is meaningfully better than the 19 to 20 months expected for SOC (as will be shown shortly), we can infer that this is because of a strong therapeutic benefit from DCVax-L plus SOC.
Comparing mOS Results from the Optune Phase 3 Trial with Blinded Data from the DCVax-L Trial
In my recent report on Novocure, I discussed the clinical trial results for Optune in some detail. You might want to refer to that report before going further. The next table compares mOS from the DCVax-L trial to the Optune phase 3 trial.

Here are my key takeaways:
- In its key pivotal trial. Optune added to SOC produced an mOS from time of surgery of about 24.4 months versus 19.5 months for SOC. This was an improvement of 4.9 months.
- Blinded data from the DCVax-L trials for all 331 patients showed an mOS of 23.1 months from time of surgery which compares to mOS of 19 to 20 months seen in other trials of newly diagnosed glioblastoma. Hence, the blinded results show an improvement of 3 to 4 months.
- I think that the mOS results for the blinded data would be sufficient for approval for DCVax-L. However, it seems highly likely that DCVax-L plus SOC will be meaningfully higher than 23.1 months unless the SOC patients in the trial achieved a never before seen mOS results which is extremely unlikely. I can’t really guess how much higher mOS for DCVax-L added to SOC might be, but it probably will be as good or slightly better than that achieved by Optune.
Generally, oncologists consider an improvement in mOS of 4.0 months in an aggressive cancer like newly diagnosed glioblastoma to be a significant advance. In 2005, the chemotherapy drug temozolomide was approved as part of SOC based on a 2.5 month improvement in mOS Taking all of this into account, I think it is reasonable to hypothesize DCVax-L has an excellent chance of showing a therapeutically meaningful improvement in mOS that will result in approval.
Comparing Survival Outcomes in the Two Trials
The next table compares the percentage of patients alive at two, three, four and five years. Unlike with mOS, it is not feasible to make an “apples to apples” basis from time from surgery. However, I don’t think it makes that much of a difference in this case given that the time intervals are in years rather than months. In addition to the data from the Optune and DCVax-L trials, I have also included survival data from the Stupp trial in 2005 that defined current standard of care. Like the Optune data, this is from time of randomization rather than time of surgery.

The above table shows that the two and three year survival for the blinded data from the DCVax-L trial was comparable or slightly better than the data for Optune plus SOC and dramatically better than for the SOC arm of the Optune trial and the SOC arm in the Stupp trial. Per my argument on mOS data, I would expect survival results for DCVax-L plus SOC to be meaningfully better than what is seen with the blinded data. The above table indicates that both Optune and DCVax-L have therapeutically important survival tails.
Survival and mOS Results Seen with SOC in Other Trials in Newly Diagnosed Glioblastoma
A reasonable question is how reliable is the data on standard of care. Later in this report (for the dedicated reader) I discuss some of the complexities of the DCVax-L trial that may make difficult a clean comparison between the effects of DCVax-L added to SOC versus SOC alone. If so, the FDA may need to put significant weight on results seen with SOC in other trials in newly diagnosed glioblastoma. There is a great deal of data from other trials that assess mOS for the SOC control arm of other trials of investigational drugs in phase 3 trials. As can be see below, the SOC results are very consistent across trials:
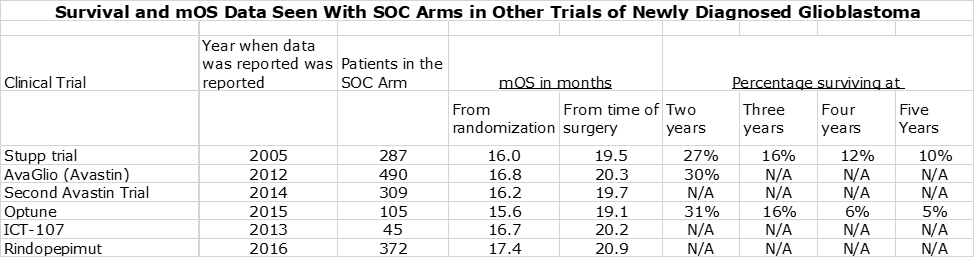
The table above indicates that mOS for these six trials showed a tight range of 15.6 to 17.4 months as measured from the time of randomization and about 19.1 to 20.9 months from time of surgery. We probably want to consider the 17.4 months from the rindopepimut trial as an outlier, because it enrolled only patients with the EGFR mutation. Despite the reluctance of the FDA to compare results from one trial to another, I think that the large number of patients (1,608) and the tight results from one trial to another will give the FDA considerable comfort that SOC in newly diagnosed glioblastoma delivers an mOS of 19 to 20 months from time of surgery.
This argument is important because the blinded data of all 331 patients in the DCVax-L trial which combined those patients started on DCVax-L plus SOC and those started on SOC. This showed an mOS of 23.1 months from time of surgery. If the SOC patients performed in line with historical results then the DCVax-L patients must have done better than 23.1 months and produced a very meaningful therapeutic benefit.
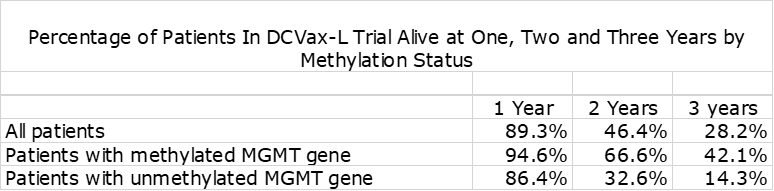
Methylated Versus Unmethylated Patients
In nGBM, one of the most important factors affecting survival is the status of the promoter region of the MGMT gene. If it is unmethylated, it produces an enzyme that blocks the effect of temozolomide resulting in meaningfully lower mOS This is shown in the following table. The outcome for Optune added to SOC is comparable to blinded data from the DCVax-L trial.

The blinded data from the DCVax-L phase 3 trial shows the dramatic effect on survival at two and three years from time of surgery.
Issues for the FDA in Evaluating the Results from the Phase 3 Trial of DCVax-L
Considerations for the FDA
In its decision to approve a new drug, the FDA is charged with determining whether the new therapy results in a therapeutically meaningful improvement in comparison to the current standard of care. In oncology, the majority (although not all) of clinical trials compares one group of patients given a new drug added to SOC versus another group given just SOC. The results in each arm of the trial are compared by determining and prospectively specifying therapeutically meaningful measurements (endpoints). The results are then analyzed based on a rigorous statistical analysis to determine if we can have a high level of confidence that the new drug does improve outcomes relative to SOC.
The FDA prefers that the design of the clinical trial allows such a determination to be made by data from the trial. The agency is cautious in drawing conclusions by comparing results in one trial to other similar trials; nevertheless, it can still be an important factor. However, as I have previously shown, SOC results from other trials do give a firm understanding of the survival for nGBM patients that can be expected from SOC and could carry some meaningful weight in the FDA’s decision making.
In the case of approval of oncology drugs, the agency is laser focused on whether a drug provides a survival benefit and the duration of that benefit. If the still to be unblinded results from the DCVax-L phase 3 trial leads to a regulatory filing, the agency will heavily focus on the essential question of whether adding DCVax-L to SOC results in a therapeutically meaningful improvement in survival over SOC alone.
Design of Phase 3 Results in Complexities in Determining Survival Advantage of DCVax-L
The DCVax-L phase 3 trial may have some complexities in trying to compare the effect of DCVax-L added to SOC versus SOC in the trial. Here’s why. The design of the DCVax-L resulted in 331 patients being randomized about 2:1. This means that about 232 initially were given DCVax-L plus SOC and about 99 initially received SOC. Patients who progressed on SOC were given the option to receive DCVax-L so that at the end of the trial, about 90% of patients (298) had received DCVax-L. This indicates that about 69 patients who began on SOC later received DCVax-L. Roughly 30 patients received only SOC. Another factor to consider is that patients who began on DCVax-L plus SOC and saw their cancers progress could choose to remain on DCVax-L. We then have the following patient groups:
- Started on DCVax-L plus SOC and completed the therapeutic regimen
- Started on SOC and were given DCVax-L when their cancer progressed
- Only received SOC
- Started on DCVax-L and remained on the drug even if their cancer progressed
At this time, we don’t know how many patients fall in each group. Further complicating the situation is that in most trials (and probably in this one) some patients drop out of the trial or are lost to follow-up in one or more of the above groups. This could make for a complicated statistical analysis plan (SAP). As I discussed in my prior note, after a considerable amount of work, the draft SAP was finalized last fall.
Some skeptics have suggested that that there is effectively no control group. Because of the cross over design, 90% of the patients in the trial received DCVax-L at some point and there were only about 30 patients who received just SOC. Because of the small number of patients who only received SOC, skeptics argue that a meaningful comparison may not be possible. It is likely also the case that patients in this roughly 30 patient group were those who did not see their cancer progress and most benefitted from SOC or patients who elected not to receive DCVax-L when their cancer progressed. It could also include those who rapidly progressed and died.
Proposed New Guidelines Are Very Positive for DCVax-L
This report is focused on comparing the blinded results of the DCVax-L trial with the results from the phase 3 trial that led to the approval of Novocure’s medical device Optune in nGBM. However, it is important to juxtapose this with proposed guidelines from the FDA advising companies on what type of trial design and clinical data will now be acceptable to the FDA in considering regulatory approval. On December 19, 2019 the FDA released for comment proposed guidelines called Demonstrating the Substantial Evidence of Effectiveness for Human Drug and Biological Products. Here is the link.
Aggressive short sellers of NWBO have claimed that the FDA will not approve DCVax-L because of flaws in the trial design. They reference the general agency guideline that approval should be based on two well controlled phase 3 trials which compare a new drug to placebo or effective therapy. Because Northwest executed only one phase 3 trial, the shorts argue that NWBO will have to do a second confirmatory trial. This would be devastating. The new guidelines dismiss this argument. They state that in the case of life threatening diseases with unmet medical need like nGBM that the FDA will consider one well controlled trial as adequate for approval.
Actually, this is already how the FDA is currently operating. It has become common for the FDA to approve drugs on the basis of just one trial. Optune was approved on the basis of just one phase 3 trial. The CAR-T drugs Kymriah and Yescarta were approved for r/r DLBCL on the basis of open label, phase 2 trials that enrolled 115 and 112 patients respectively. Interestingly, the endpoint of these two trials was objective response rate (tumor shrinkage). At the time of approval, there was no meaningful data on survival. The phase 3 trial of DCVax-L initially enrolled 232 patients on DCVax-L plus SOC and very importantly, there is strong data on survival as the last patient was enrolled over four years ago. It is a much more meaningful data base than that which led to the approval of Kynmriah and Yescarta. The mOS of r/r DLBCL before the CAR-T drugs was about eight months and the mOS of nGBM with SOC is about 19 to 20 months. Both would seem to meet the FDA definition of life threatening, highly aggressive cancers. We can easily dismiss the short sellers’ contention that having only one phase 3 trial of DCVax-L is inadequate for approval.
The short sellers also argue that the FDA will not approve DCVax-L because it does not have a large enough SOC group against which to compare results for DCVax-L plus SOC. As previously discussed, the trial allowed patients who progressed on SOC to then receive DCVax-L. As a result, only about 30 patients in the trial received just SOC and never received DCVax-L. This compares to about 99 patients who initially received SOC. The short argument is that this small sample of SOC patients will result in a p value much higher than the p≤0.05 generally required for approval. However, the proposed guidelines state that in the case of a life threatening disease like nGBM, FDA will consider historical results from other trials. In this report, I have identified six trials in which 1,608 patients were given just SOC. The mOS in these patients fell in a tight range of 19 to 20 months. The proposed guidelines indicate that NWBO can statistically compare DCVax-L plus SOC results in the phase 3 trial to these historical results. This is huge.
Looking down the road, this new willingness to accept historical controls also augurs well for future trial of DCVax-L in other solid tumors. The FDA also says that if mechanism of action has been established as hopefully will be the case in nGBM, that it will take this into account as trials are done to expand the label into other indications; i.e. other solid tumors. This could result in the approval of DCVax-L for numerous other solid tumors on the basis of small phase 2 trials like those that led to the approval of Kymriah and Yescarta. This is equally huge.
Tagged as DCVax-L blinded phase 3 results, DCVax-L data versus Optune, Northwest Biotherapeutics Inc. + Categorized as Company Reports, LinkedIn
Thanks for the article. With the new draft guidelines for FDA, will this make NWBO want to wait on unblinding until after the draft is adopted or does the draft only need to be adopted before DCVax-L is considered by FDA for approval? Is that when the NDA or BLA is filed? Could this draft be in part what NWBO is/was waiting for before proceeding with data lock?
In actuality, the FDA is already largely following these guidelines. Refer to my comments on the CAR-T products in my report. Still, formalizing them is important. The timing on formalization won’t affect the filing, in my opinion.