



Northwest Biotherapeutics: Why I Believe That the Phase 3 Trial of DCVax-L in Newly Diagnosed Glioblastoma Patients Has a Very Good Chance for Success (NWBO, Buy, $0.17)
Overview
This report is an extensive update on almost everything I know about the DCVax-L phase 3 trial and is over 20 pages long. The catalyst for this report was the recent information on the still blinded phase 3 trial presented in June at ASCO. It will take dedicated investors a great deal of time to read this report in its entirety. For a quick overview, the Key Points section summarizes my key conclusions and can be quickly read. The other parts will take more time and effort so that you may want to read just one of these sections at a time. Here is the organization of the report.
- Key Points lists my most important conclusions and hypotheses.
- New Information from ASCO
- Encouraging Open Label data from the Information Arm of the Phase 3 Study and the Phase 1/2 Studies
- Background Information for Help in Understanding the Phase 3 Trial
- Thoughts on Potential FDA Interpretation of Phase 3 Trial
- Examples of the Long Term Survival Tail for the Checkpoint Inhibitor Opdivo
Key Points
- I continue to believe that the phase 3 trial of DCVax-L in newly diagnosed glioblastoma has a very good chance for success. The trial remains ongoing and blinded.
- Key opinion leaders involved in the trial have stated that patients appear to be living much longer than would be expected if they were treated only with current standard of care.
- The trial remains blinded but new information on the phase 3 trial presented at ASCO adds supports to the hypothesis that DCVax-L is providing an impressive survival benefit over and above standard of care.
- This long term survival hypothesis is further reinforced by open label data which comes from two other data sets: (1) 25 patients treated in the information arm of the phase 3 trial and (2) 20 patients treated in the phase 1/2 trial. Both data sets showed impressive survival benefits as compared to standard of care and long term survival tails that compare favorably to those seen with checkpoint inhibitors.
- Results from the two studies just cited suggest that with DCVax-L, the difference in the percentage of patients who remain alive for a long time period (say two or more years) as compared to the percentage of patients who remain alive on SOC at that same time point (the survival tail) is comparable to that seen with checkpoint inhibitors (Opdivo, Keytruda, Tecentriq) in other aggressive solid tumors such as advanced cases of non-small cell lung cancer and melanoma. It is the long term survival tail that has excited key opinion leaders about the checkpoint inhibitors.
- The design of the phase 3 trial is such that patients whose cancers progress on standard of care are switched to DCVax-L. Also, patients who progress on DCVax-L remain on DCVax-L because trials with other immune therapies like the checkpoint inhibitors have shown survival benefits even after the cancer has progressed. This is all done in a blinded fashion.
- The median time for progression for standard of care is about seven months so that many patients on SOC could have been quickly switched to DCVax-L. It was reported at ASCO that 90% of the patients in the trial were on DCVax-L. This is very close to being a blinded single arm study and could explain why patients appear to be living longer than would be expected with SOC.
- We know that the trial was randomized 2:1 in favor of DCVax-L so that 67% or 221 of the 331 patients enrolled in trial revieved only DCVax-L. By simple arithmatic, we can calculate that 23% or 76 of the patients first received standard of care and were switched to DCVax-L and that 10% or 34 received only standard of care. This cross over design does not affect the statistical analysis of mPFS but it does require a special analysis for mOS which could create some ambiguity in ferreting out the effect of DCVax-L. I understand there is a statistical method for this type of analysis.
- Because of this aspect of the trial, I think that the FDA may place great weight on the length of time that patients survive as compared to historical SOC results determined in other trials. This would be in addition to the analysis of the two primary but independent endpoints of mPFS and mOS.
- One of the strong attributes of DCVax-L is its safety profile. The most frequent side effects are a fever after injection that can be treated with Tylenol and mild pain and irritation at the site of intradermal injection. Out of more than 2,000 intradermal injections given to over 400 patients, there have only been 7 patients who experienced serious adverse events that investigators deemed to be related or possibly related to the DCVax-L treatment. Five of these were seizures and it is important to note that because GBM is growing rapidly in the brain that the disease itself can cause seizures. Because safety can be as important to a drug as its efficacy, this is a major plus for DCVax-L.
- The trial is likely to continue for an indeterminate time beyond July at which time one of the minimum stopping thresholds for the trial of 233 deaths is likely to be reached. This is because of a statistical technique called censoring. With censoring, all patients still alive in the trial would not be included in the analysis of mOS, one of the two primary endpoints in the trial. In the case of the DCVax-L trial, the 100 patients still alive would be dropped from the analysis and the calculation would be based on the 231 patients who have died. However, some of the patients who responded best to DCVax-L are likely to be included in this group of 100. Because of this, the Company has a compelling reason to let the trial run longer in order to to allow the data to mature further.
- The FDA has shown much greater flexibility in approving new cancer drugs in the last few years. It has approved the checkpoint inhibitors Opdivo and Keytruda on the basis of small studies with end points of objective response or mPFS with no data on survival in some aggressive cancers. The data set for DCVax-L will be large and will have much data on survival. In the event of ambiguities in the two separate primary endpoints of mPFS and mOS, the long term survival that I am hypothesizing might be compared to historical SOC results and could be a key factor in the FDA’s decision on approval.
- The Company is burning about $1.5 million per month. They are financing this burn by raising small amounts of capital that usually provide only a few months of cash. The goal is to minimize the amount of dilution until the topline results of the phase 3 trial are available. This could potentially involve one, two or more such small raises.
- If my hypothesis about results in the phase 3 trial is correct, it should lead to approval. DCVax-L could be a major medical advance and commercial blockbuster, possibly comparable to the checkpoint inhibitors. I am obviously standing virtually alone in this opinion as evidenced by the $0.17 penny stock share price and micro-cap valuation of $33 million placed on the company. If my view were the consensus, the stock would likely have a market capitalization of $1 billion or more.
- The trial remains ongoing and blinded. Neither the Company nor investigators will know how the results for DCVax-L added to SOC compares to SOC alone until the trial is ended; the data base is locked and scrubbed; and statistical analyses are performed. The arguments I present in this note are suggestive, but by no means conclusive evidence that the trial will be successful.
Acronyms Used Extensively in this Report
- GBM: Glioblastoma multiforme is stage 4 brain cancer
- SOC: The standard of care in newly diagnosed GBM patients is surgical resection followed by treatment with radiation and the chemotherapy drug temozolomide
- PFS: Progression free survival is the time from when the surgery is performed until the tumor begins to regrow
- mPFS: The time at which the cancers of half of patients in a clinical trial have progressed
- OS: Patients who remain alive from the time of surgery
- mOS: The median time at which half of patients in a clinical trial are still alive
New Information from ASCO
Information from ASCO is Encouraging, But Not Conclusive
Dr. Marnix Bosch, Chief Technical Officer of Northwest Biotherapeutics, gave a presentation at ASCO on June 5th that provided new information on the phase 3 trial of DCVax-L in patients with newly diagnosed GBM. In his presentation, he showed for the first time a graph of patient enrollment in the trial which indicated that the median time of enrollment was May, 2014, slightly over 3 years ago. This is a very long running trial in which the first patient was enrolled in June 2008 and the last in November 2015. This enrollment timeline is shown below:
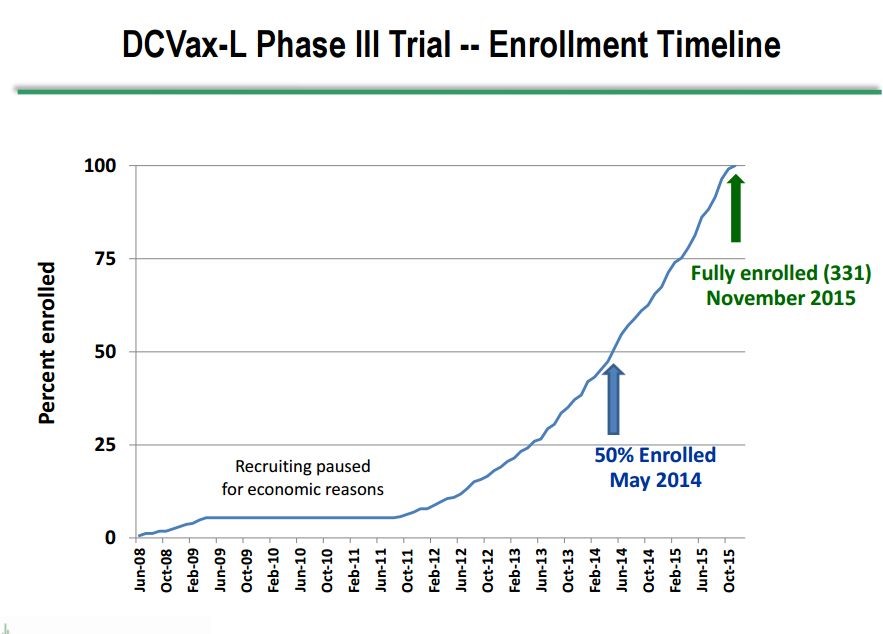
Key investigators in the phase 3, DCVax-L trial have stated that patients appear to be living longer than would be expected if they had been treated only with SOC. An encouraging sign of longer patient survival is that the trial was anticipated to end some months ago, but it remains ongoing because the rate of deaths has been slower than expected. The new blinded data released at ASCO further strengthens the hypothesis that we are seeing longer patient survival in the trial due to DCVax—L. Here is what we learned from Dr. Bosch at ASCO.
- The median time of enrollment in this 331 patient trial was May 2014, which was 37 months ago as of June. It is important to understand that enrollment in the trial usually starts about three months (actually 13 weeks) after surgery. This means that for patients still alive the length of survival is equal to the time from when they were enrolled plus three months. Keep this three month difference between enrollment and survival in mind.
- The first patient was enrolled in June 2008 and the last in November 2015.
- There are 100 patients still alive in the trial.
- 90% of patients in the trial received DCVax-L. Two thirds of the 331 patients were initially given DCVax-L. It is very important to understand that the trial was constructed so that anyone whose cancer progressed then would be given DCVax-L. This meant that patients who progressed on SOC would be switched to DCVax-L and those who progressed on DCVax-L would continue on DCVax-L. This feature allowed the trial to remain blinded, but in actuality, it was close to being a single arm trial.
- The reason for keeping patients on DCVax-L after they have progressed is that we have seen in trials of other immune therapies that meaningful survival benefits have been seen in patients even after their cancer progressed.
- There are two separate and independent endpoints for the trial-PFS and OS. The minimum stopping threshold for PFS is 248 progression events and that has been reached although Dr. Bosch mentioned in his presentation that further analyses will also be done as part of completing the trial. . The minimum stopping threshold for OS is 233 deaths and as of the ASCO presentation only 231 deaths have occurred. The death rate has slowed to about 2 per month so that the 233 event rate could be reached in July. Importantly, the trial is not likely to be stopped then, but more on this later.
Some Thoughts on the 100 Patients Still Alive
Using assumptions as explained below, I estimate that only 22 of these 100 patients would be expected to be alive if they had received only SOC. It is highly likely that 78 of these 100 patients have experienced a survival benefit due to DCVax-L. It is also important to understand that this does not mean that only these 78 patients benefitted from DCVax-L. As I explain shortly, it is quite probable that some of the other 231 patients who have since died experienced a significant survival benefit before dying. There is just no way to attempt to measure this using information currently available. Here are the assumptions that I used to reach the estimates just cited.
- We won’t know precisely how many of these 100 patients were on DCVax-L until the trial is unblinded. However, it seems pretty reasonable to assume that about 90% were on DCVax-L during the trial consistent with the percentage for all 331 patients enrolled. This is essentially a single arm trial with results driven by DCVax-L.
- We know that the very last person enrolled in the trial was enrolled 19 months ago so that if this person is still alive and part of the 100 they have survived 22 months since surgery. All other patients in this group of 100 would have survived longer than this.
- It would be extremely helpful to know when all of the patients were enrolled and when they died. We could then calculate the length of survival for each and compare to the expected survival if they had been treated with SOC, but this can only be determined when the trial is unblinded. Barring this, maybe we can find a reasonable estimate of the range of survival for the 100. We know that the lower end of the range is 22 months of survival but what is a good estimate of the upper end?
- Let me suggest an artificial but still reasonable way of approximating the upper end of the range. Let’s make a simplifying assumption that all 165 or so patients who were enrolled before the median enrollment data of May 2014 have died. This would mean that all of the 100 patients still alive were enrolled between May of 2014 and November of 2015. With this assumption, as of June the survival range would be 22 to 40 months for this group of 100 patients. To the extent that some of these patients were enrolled before May 2014, the 40 month estimate for the upper end of the range would be low.
- Clinical trial data suggests that about 36% of GBM patients treated with SOC would be alive at 22 months after surgery and about 8% would survive for 40 months. If these 100 patients had been treated with SOC and if we assume a standard distribution curve for enrollment we can estimate how many of the 100 patients would be expected to survive if they were treated only with SOC. We can simply average the 36% expected survival at 22 months with 8% at 40 months and estimate that about 22% of these 100 patients would be alive. This would indicate that only 22 of these 100 patients would be expected to be alive if they had received only SOC. If so, DCVax-L has provided a survival benefit to 78 of these 100 patients over and above SOC.
You can criticize my analysis in many different ways, but I think that this gives a rough but reasonable estimate of the potential benefit of DCVax-L and that we are seeing an impressive length of survival in these 100 patients which would not be expected if they had received only SOC. Since it is highly probable that 90% or more of these patients received DCVax-L, it is very reasonable to hypothesize that DCVax-L provided this very meaningful survival benefit over what would be expected with SOC.
I want to emphasize that this does not mean that only these 100 patients may have experienced a survival benefit better than that expected from SOC; some of the other 231 patients who died during the trial could have also benefitted. Let me give a hypothetical example to illustrate this. Let’s assume that a patient was treated with DCVax-L in 2010 and lived until 2016. This would be an extraordinary result in prolonging the patient’s life by nearly five and one-half years beyond what would be expected with SOC. However, they eventually died and would not be part of the 100 patients remaining alive. There could be potentially many patients like this hypothetical example who achieved a significant increase in survival relative to SOC but ultimately died. My analysis only deals with the 100 patients still alive and makes no estimate of benefit for the other 231 patients who may have died after achieving a meaningful survival benefit.
The trial remains ongoing and blinded. Neither the Company nor investigators will know how the results for DCVax-L added to SOC compares to SOC alone until the trial is ended, the data base locked and scrubbed and statistical analyses is performed. The arguments I present in this note are suggestive, but by no means conclusive evidence that the trial will be successful.
Why the Trial Could Continue for Awhile
Let me switch to another point. The trial has one primary endpoint of progression free survival (PFS) and a second endpoint of overall survival (OS). Very importantly, As Dr. Bosch noted in his presentation at ASCO, these are independent and not co-primary endpoints. They are statistically powered for significance at 248 progression events and 233 deaths in accordance with trial assumptions about how DCVax-L will compare to SOC. The 248 progression events were reached some time ago and the 233 deaths could occur in July. Some investors are confused as to why the trial was not stopped when 248 progression events were reached and are even more confused by the probability that it will not be stopped at 233 deaths. So what is going on?
There is an extremely important reason for letting the trial continue. When the trial is stopped, and the mOS analysis is performed, patients who are still alive at that time will be censored or dropped from the analysis. This would mean that the 100 patients who are still alive would not be considered in the mOS statistical analysis. These are very likely the patients who are most benefitting from DCVax-L.
When the trial is stopped, those DCVax-L patients still alive would be censored (not included) from the data set used to determine mOS. This could result in significant negative bias against DCVax-L as the 100 patients still alive could very well include some or many of the best responding patients who are still alive. Because of this, NWBO, has good reason to keep the trial ongoing and allow the date to further mature.
Will DCVax-L Have a Long Survival Advantage or Tail?
DCVax-L is an immune therapy and we have learned from experience with other immune therapies like the checkpoint inhibitors Opdivo and Keytruda that their most compelling aspect is that in many cases there is a survival tail in which a greater percentage of patients treated are alive at time points of one, two, three years and more than with previous SOC. Just to illustrate the effect, it might be the case in an aggressive cancer comparable to GBM that the time of survival at some time point is 25% for patients given checkpoint inhibitors versus 10% for previous SOC. This long term survival advantage or tail is what has excited key opinion leaders. Later in this report I discuss three separate trials of Opdivo in different cancers in which the percentage of patients surviving on Opdivo was 13%, 15% and 18% more at two years than the percentage of patients surviving who were treated previous standard of care.
In the next section I discuss the results for 25 patients in the information arm of the phase 3 trial in which 40% of patients treated with DCVax-L are alive at 35 months versus an expectation of 15% of patients who would be expected to be alive with SOC. The difference of 25% compares very favorably to the 15% to 20% difference that is frequently seen with Opdivo and other checkpoint inhibitors.
I also discuss 20 patients in the phase 1/2 trial in which 33% of patients treated with DCVax-L have survived for four years or more versus an expectation of 8% with SOC. The difference is 25% comparable to what was seen in the information arm patients.
The long time span over which the phase 3 trial has been conducted will give a very meaningful insight into the survival tail of DCVax-L. In the event that the increase in percentage of patients treated with DCVax-L are alive at say a two, three or four year time point exceeds expected survival for patients treated with SOC by 25% as has been seen in the information arm and phase 1/2 trial, this would be better than what we have seen with Opdivo in advanced non-small cell lung cancer and melanoma. This would be an amazingly positive result. However, let me caution that it is often the case that results from large phase 3 trials are less robust than results seen in smaller trials.
Extremely Encouraging Open Label Data from the Information Arm of the Phase 3 Study and the Phase 1/2 Studies
Information Arm of the Phase 3 Trial
There are two sets of unblinded data for DCVax-L that are extremely encouraging. The first comes from the Information Arm of the phase 3 trial. There were 51 patients who were screened for the phase 3 trial and for whom DCVax-L treatments were prepared. Forty six of these patients showed signs of disease progression before they could be enrolled, so they essentially would have failed the primary endpoint of disease progression before they even received DCVax-L. There were also five patients for whom images were not available. Altogether there were 51 who were not enrolled in the trial, but they were treated and followed as if they were enrolled.
Investigators determined that there were 20 patients who upon follow-up examinations were judged to be rapid progressors whose cancer had recurred. One other patient was determined to be a pseudo-progressor. These patients initially appear to have a progression of the tumor but actually the tumor appears to be progressing because of inflammation caused by an immune response to the tumor. These are usually the best responding patients. Finally, there were five patients who could not be classified because of lack of scans.
This left 25 patients whose cancers progressed rapidly but less so than for the 20 patients. The difference was that the 20 patients showed disease progression at the first baseline scan and then at the follow-up scan about 8 weeks later. The 25 patients showed disease progression at either the baseline scan or the next follow-up scan but not both. All of these determinations were made by blinded assessment at a central radiology lab by blinded investigators. Northwest Biotherapeutics had no involvement. This is shown below:
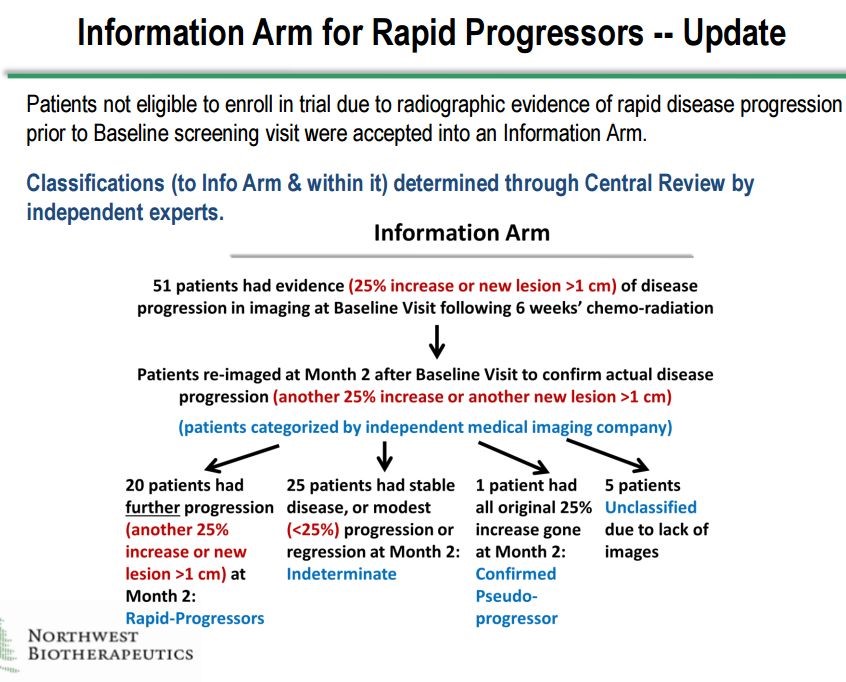
These 25 patients give a potentially valuable insight into what may be going on in the phase 3 trial. They are sicker patients whose cancer progressing sooner and more rapidly than those who are enrolled in the phase 3 trial. One might expect that these patients would respond less well so that results in this group gives a potential insight into the results for the phase 3. Here is a waterfall plot showing impressive survival results for these 25 patients.
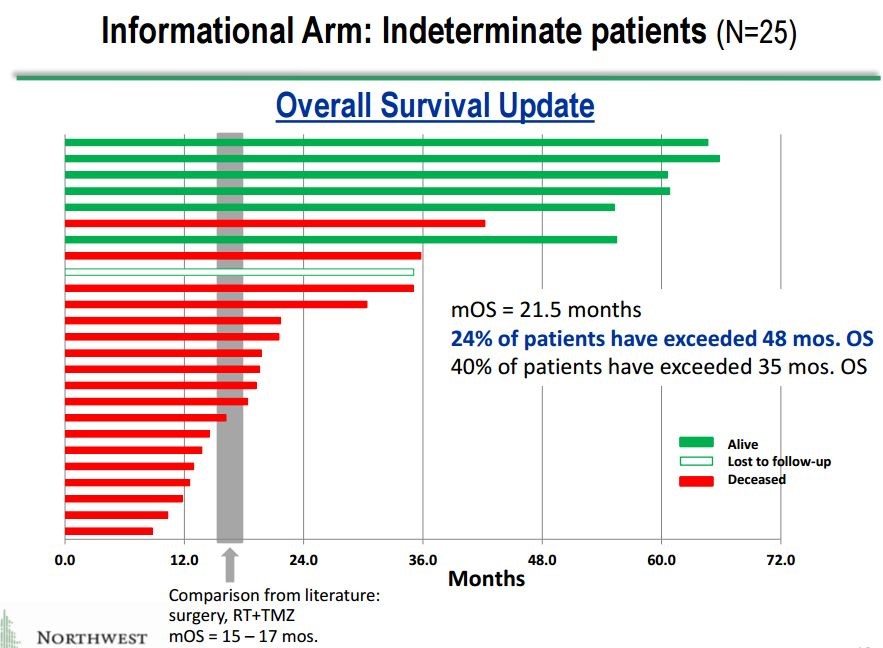
This is a relatively small group of 25 patients, but the results are extremely impressive.
- The mOS is 21.5 months which is a 4.5 month improvement over the 16.0 months expected with SOC in newly diagnosed GBM. However, as I previously noted these are probably sicker patients than those enrolled in the phase 3 trial so that we might hope for better results in the phase 3 trial? Still, in cancer drug development, a 4.5 month improvement is considered a major advance.
- Even more intriguing is the suggestion of a very meaningful long term survival tail. In the previous section in which I discussed the long term survival tails for Opdivo in melanoma and non-small cell lung cancer, I noted that at about two years that based on a percentage of patients still alive roughly 15% to 20% more Opdivo treated patients were still alive as compared to SOC.
- 40% of patients treated with DCVax-L remained alive at 35 months which compares to 15% that would be expected for SOC. This indicates that 25% to 25% more of the total DCVax-L patients treated are surviving at three years than would be expected with SOC.
- At four years, 24% of patients are surviving versus an expected 8% for SOC. This indicates that 16% more of the total DCVax-L patients treated are surviving at four years than would be expected with SOC.
This suggests a survival tail that compares extremely well to that seen with Opdivo in advanced non-small cell lung cancer and melanoma. If this is predictive of final results in the phase 3 trial, DCVax-L will be a major therapeutic advance in the treatment of glioblastoma multiforme.
These 25 patient and 20 patient data sets may not seem like a lot for the FDA to consider. However, I would point out that FDA assigned breakthrough drug status to Novartis’ CAR-T product CTL-019 based a single arm study in only 51 patients. I might add that this was also based on a primary endpoint of tumor shrinkage at six months and about 30% of patients achieved a complete response. There is not yet any meaningful data on survival. It seem to me that the DCVax-L data from the information arm compares very favorably (to me it looks more impressive) than the CTL-019 data. There is actual survival data, not just tumor shrinkage.
More Very Encouraging Open Label Data from the Phase 1/2 Trial
The basis for undertaking the phase 3 trial was very encouraging data from 20 newly diagnosed patients in phase 1/2 trials. These trials also treated 14 patients with recurrent GBM and 5 with lower grade gliomas, but we should only focus on the 20 patients with newly diagnosed GBM which is the same criteria as for those enrolled in the phase 3 trial. Obviously, there was no control group but results were compared to historical results for standard of care and also matched controls. These were newly diagnosed GBM patients who presented at about the same time but were treated only with SOC. Results are shown below:
IMAGE 4

The results were extremely impressive:
- DCVax-L increased mPFS by about 16 months over matched concurrent controls and by about 17 months over historical SOC. In the phase 3 study, the powering is such that the primary endpoint on mPFS would be reached with a four month improvement.
- DCVax-L increased mOS by about 17 months over matched concurrent controls and by 15 months over historical SOC. Remember that a 4.5 month increase in mOS for a cancer drug is generally considered a major advance.
- Thirty three percent of patients were alive at 4 years versus an expectation of about 8% for patients treated with SOC. This indicates an extremely impressive survival tail at four years of 25%. Remember that Opdivo delivers something like 15% to 20% in advanced non-small cell lung cancer and melanoma at two years.
- There are 27% of patients alive at six years or more and two patients have survived more than 12 years. Were these patients effectively cured with DCVax-L?
Background Information for Help in Understanding the Phase 3 Trial
Background on Glioblastoma Multiforme and DCVax-L
Northwest Biotherapeutics is conducting a large, randomized phase 3 trial of DCVax-L in newly diagnosed GBM. This trial is extremely important from both a scientific and commercial standpoint. GBM is a very aggressive cancer in which 50% of patients treated with SOC die about 16 months after surgical resection. Because the cancer is growing rapidly within the confines of the skull, it impinges on normal functions of the brain playing havoc with quality of life. There have been no drug advances in the treatment of GBM since 2005 when the Stupp trial established current SOC although in 2015, the FDA approved a medical device for the treatment of GBM. These have been the only drug/ device advances in survival of GBM patients during the last 12 years.
DCVax-L is a dendritic cell vaccine that takes a completely novel drug approach that could be paradigm shifting. The phase 3 trial of DCVax-L should reach conclusion this year, but more exact timing is uncertain. Until the blind is broken and data from the trial is analyzed no one really knows what is going on in the trial. Still, there are some tidbits of information that offer some insight.
In order to gain approval DCVax-L must provide a meaningful improvement in survival over current standard of care. So what is the expected length of survival for GBM patients treated with SOC? To determine this, I looked at the clinical results from four well conducted, randomized trials in GBM in which one arm of the trial was comprised of patients treated with SOC. These were:
- In 2005, the Stupp trial defined the current SOC. It compared surgery followed by radiation and the chemotherapy drug temozolomide combined to surgery followed by just radiation. Temozolomide plus radiation improved mOS to 14.6 months from 12.1 months and established a new standard of care and for the last 12 years SOC has remained unchanged. In the Stupp trial, there were 287 patients who received temozolomide plus radiation
- Roche conducted the AvaGlio trial which compared Avastin added to SOC with just SOC. The trial failed to show any improvement in mOS in the Avastin arm and it was not approved. There were 480 patients in the SOC or control arm of the trial that provided data on how SOC performed.
- Roche conducted a second trial similar to AvaGlio which also failed. There were 309 patients in the control or SOC arm of that trial.
- The latest data on SOC comes from the trial of Optune, a medical device which was approved for the treatment of newly diagnosed GBM in 2016. In this trial Optune plus SOC was compared to 105 patients treated with SOC.
These four trials analyzed data from 1,181 patients treated with SOC. The results across these four trials was reasonably consistent. It indicates that 50% of patients survive somewhere between 14.6 and 16.8 months (mean of 15.8 months) and that about 30% survive for 24 months. This is summarized in the following table.
| Data from Randomized Clinical Trials Showing Length of Survival of GBM Patients Treated with SOC | |||||
| Year when data was reported | Patients treated with SOC | mOS- Time at which 50% of patients remain alive (months) | Patients alive at 24 months (%) | Patients alive at 60 months (%) | |
| Stupp trial | 2005 | 287 | 14.6 | 26 | |
| AvaGlio | 2012 | 480 | 16.8 | 30 | |
| Second Avastin trial | 2014 | 309 | 16.2 | ||
| Optune trial | 2015 | 105 | 15.6 | 29 | 5 |
Only the Optune trial provided data beyond two years and indicated that 5% of SOC patients were alive at five years post-surgery. I next did a literature search to try to find data showing survival beyond two years. I found a meaningful number of studies which were not randomized and as rigorously conducted as the four studies just cited. One of the better summaries that I looked at can be found at this link. Based on this additional work, I filled in the gaps with estimates of survival at three, four and five years as shown below:
| Percentage of Patients Alive After Surgery Who Received Standard of Care, According to Literature | |
| Time following surgery | % of Patients Alive |
| 12 months | 67% |
| 16 months | 50% |
| 24 months | 30% |
| 36 months | 10% to 15% |
| 48 months | 5% to 8% |
| 60 months | ≤5% |
Some Investors are Confused about the Statistical Plan for the Trial
There is some confusion about the statistical plan for the phase 3 trial and in some cases this has led to very flawed interpretations. One Twitter critter renowned for his unwavering criticism of every aspect of DCVax-L and the phase 3 trial is actually claiming that the trial has failed. To understand how ludicrous this observation is, it is necessary to understand the key aspects of the trial.
The trial has a primary endpoint of progression free survival (PFS) and a secondary endpoint of overall survival (OS). These are separate, not co-primary endpoints. They are statistically powered for significance at 248 progression events and 233 deaths in accordance with trial assumptions about how DCVax-L will compare to SOC.
In the trial, NWBO has previously disclosed that the 248 progression events have occurred, although that will be subject to further reviews for accuracy as part of the data lock process as will all of the other data points. According to Dr. Bosch, we are now two events away from reaching 233 deaths. This has created confusion among investors. Some had thought that the study would be stopped and analyzed when 248 progressions occurred and certainly would be stopped after 233 deaths. The Twitter critter focused on the point that the 248 PFS events goal was reached. He maintained that at that point the trial should have been stopped and the results analyzed. Even though the trial is blinded, he maintains that NWBO management somehow knew that the data showed the trial had failed, but for some reason is keeping the trial ongoing
The Twitter critter and investors who listen to his analysis are missing a critical element of this study design. The 248 progression events and 233 deaths are minimum levels to achieve statistical significance under the design of the trial. They are not mandatory stopping points and NWBO has very strong rationale for continuing the trial beyond 248 progression events. There is now the expectation that the 233 deaths will occur in the July timeframe and that the trial will be stopped at that time and the data base would be locked and analyzed. However, this is highly unlikely to occur d for reasons I am about to explain.
It is Very Important to Understand the Role of Censoring
When the trial is stopped, and OS analysis is done, statistical methodology requires that patients who are still alive at that time will be censored (excluded) from the data set that is used to determine mOS. This could result in significant negative bias against DCVax-L, as this would exclude the 100 patients who remain alive in the trial. These could very well include some of the best responding DCVax-L patients. Because of this, NWBO, has good reason to keep the trial ongoing and let the data set mature.
Like all biopharma companies developing immune therapies, NWBO is making every effort to determine the extent of the long tail of survival for DCVax-L. The FDA’s ultimate decision on approval could be affected not only by looking at the outcomes for the primary endpoint of mPFS and the secondary endpoint of mOS but also at how many patients given DCVax-L are in the long tail, and how long the tail extends. In my opinion the survival tail could be the most critical consideration of the FDA. So what is a survival tail?
A Critical Goal of the Trial is to determine the Extent of the Long Term Survival Tail
Another significant reason for NWBO to keep the trial blinded beyond 233 deaths is that it is an immune-therapy. We have learned in recent years that a distinguishing characteristic with virtually all immuno-therapies is that a medically meaningful percentage of patients experience long term survival that sometimes approach a cure. This is generally referred to as a long tail.
In the case of checkpoint inhibitors like Opdivo, Keytruda and Tecentriq, it has been seen that in many cases 15% to 20% of patients experience long term survival over and above what would be expected with prior standard of care. It is this long term survival tail that has made these drugs such medical and commercial successes, and NWBO would like to demonstrate that DCVax-L has a similar long tail. In a later section of this report, I have described the survival tail for Opdivo in three cancer indications. It is a bit complicated but those who want to go into more depth may want to take a look.
Thoughts on Potential FDA Interpretation of Phase 3 Trial
Endpoints of the Trial Could Produce Equivocal Results but This Might Not Preclude Approval
Key opinion leaders caution that the phase 3 trial design for DCVax-L is based on traditional measures intended to determine efficacy for chemotherapy drugs. The focus of these endpoints is on tumor shrinkage and preventing progression, which may not fully capture the long term tail effect seen with immuno-therapy. The primary endpoint for this trial of mPFS may be inappropriate. In some trials of the checkpoint inhibitors, they have failed to show a benefit in mPFS while meaningfully improving mOS. Moreover, the cross over design of the trial allowing patients who progress on SOC to receive DCVax-L may confound analysis of the secondary endpoint of median overall survival.
KOLs caution that the FDA may have to approach this data with more of an open mind than just basing its decision on achieving the primary or secondary endpoints. The long term survival tail will likely be given heavy weight. There may be other measures such as sub-group analyses that might identify a prospectively defined group of patients who would meaningfully benefit from the drug. The FDA has shown flexibility in granting approvals for Opdivo and Keytruda based on small phase 2 trials without a control group in refractory tumors without evidence of an effect on survival. Indeed, Kite and Novartis are banking on approval of their CAR-T drug based only on showing meaningful tumor shrinkage in about one third of patients with r/r DLBCL and without evidence of a significant increase in survival. The Kite trial it is banking on is a small open label trial that enrolled just 80 patients and the Novartis trial enrolled just 51.
The phase 3 trial of DCVax-L is larger and more data rich than many other cancer drug trials that have led to approval. It is not unthinkable that it could be approved even if results for progression free survival and overall survival are equivocal based on statistical analysis. There would have to be other measures that suggest there is a long survival tail. The benign side effect profile of DCVax-L is a big plus if it comes to this.
What the FDA Will Consider for Approving DCVax-L?
All of the above comments are not meant to say that the FDA will ignore mPFS and mOS and focus only on the long term survival tail. The FDA will look at mPFS and especially mOS in considering approval. So how much might DCVax-L have to improve on SOC to gain approval?
To determine this we can look at trial results for the only two approvals in the last 12 years. In 2005, the Stupp trial defined the current standard of care. At the time, surgical resection followed by radiation was the standard of care. The Stupp trial compared temozolomide added to radiation. The improvement was based on improvements in mOS was and mPFS is shown in the next table.
In 2016 the FDA approved a medical device for the treatment of newly diagnosed GBM; this was Novocure’s Optune. Optune’s technology is based on the use of low-intensity, alternating electric fields (called TTFields) tuned to specific frequencies to disrupt solid tumor cancer cell division. The theory is that electric field delivered regionally, acts primarily on rapidly dividing cells so that they target cancer cells of a specific size with minimal damage to healthy cells. Optune added to SOC has shown synergistic efficacy.
These TTFields are delivered through a portable, medical device. The complete delivery system is designed to allow patients to go about their daily activities while receiving continuous cancer treatment. The therapy needs to be delivered continuously for 18 hours each day. It includes a portable electric field generator, transducer arrays, rechargeable batteries and accessories. Sterile, single-use transducer arrays (electrodes) are placed directly on the entire skull and connected to the electric field generator to deliver therapy.
The device is more than a bit awkward as it requires electrodes to be attached to the skull and a transducer and battery pack enable the electronic wave to be delivered almost continually. The patents scalp must be shaved every two to three days. You can imagine the effect on quality of life from this device. Imagine yourself walking around with a shaved scalp and electrodes on your head and a device must be worn for 18 hours per day.
The Stupp trial showed that patients treated with radiation plus temozolomide had a median overall survival of 14.6 months as compared to 12.1 months for radiation alone. The respective results for mPFS were 6.9 months and 5.8. Hence the basis for approval was a 2.5 month improvement in mOS and a 1.1 month improvement in mPFS.
The Optune trial showed that Optune added to temozolomide and radiation resulted in mOS of 20.5 months as compared to 15.6 months for SOC and the respective results for mPFS were 8.1months and 4.0. Hence the basis for approval was a 4.9 month improvement in mOS and a 4.1 month improvement in mPFS.
These two trials suggest that if DCVax-L shows something like a 3 to 5 month improvement in mOS that it is likely to gain approval.
Recent FDA Actions Suggest That Standards for Approval Are More Flexible
One thing that must be figured into investor thinking is that the FDA appears to be more flexible on clinical data that is being required for approval. The agency appears to have moved away from relying on rigid statistical interpretation of data toward looking at the preponderance of data. Certainly, in aggressive cancers like GBM it has been approving drugs based on data that would have been rejected perhaps four years ago. This could be important for DCVax-L in the event that the preponderance of the data suggests efficacy, but for whatever reason some of the statistical analysis on mPFS and mOS is equivocal or inconclusive.
My point is that clinical trials virtually never answer all of the questions we have about a drug and there are invariably ambiguities. I think that if this occurs in the DCVax-L trial, the FDA will lean heavily toward approving the drug as opposed to asking for more data. You are probably asking what possible ambiguities might occur clinical data or trial design. I can’t say other than that in almost all trials there are always such issues.
If this is the case, one of the really strong attributes of DCVax-L is its safety profile. The most frequent side effects are a fever after injection that can be treated with Tylenol and mild pain and irritation at the site of intradermal injection. Out of 2,000 intradermal injections to well over 400 patients, there have only been 7 patients who experienced “serious adverse events” that investigators deemed to be related or possibly related to the DCVax-L treatment. Five of these were seizures and it is important to note that because GBM is growing rapidly in the confined area of the skull that the disease itself can cause seizures.
The side effect profile is extremely benign relative to chemotherapy and new drugs like the checkpoint inhibitors Opdivo and Keytruda and the CAR-T therapies. All of these drugs have an incidence of 30% to 50% of serious adverse side effects. The FDA makes its decision on approving a drug on the basis of efficacy versus side effects. With its benign side effect profile, this weighs very much in factor of DCVax-L in any approval decision.
Recent Approvals Suggest FDA Has Become More Flexible on Approvals
I now want to turn to some examples of recent FDA decisions on approval.
Example 1: On May 10, 2017, Merck announced that the FDA approved the combination of Keytruda with the standard chemotherapy regimen that has been standard of care for the first-line treatment of metastatic non-squamous NSCLC, irrespective of PD-L1 expression. Under the FDA’s accelerated approval regulations, this approval was based on tumor response rate (tumor shrinkage) and progression-free survival. Continued approval for this indication may be contingent upon verification and description of clinical benefit in the confirmatory trials.
The approval was based on data from one cohort of the KEYNOTE-021 trial which was comprised of one small contingent of 123 previously untreated patients with metastatic, non-squamous NSCLC with no EGFR or ALK genomic tumor aberrations and irrespective of PD-L1 expression.
In this trial, Keytruda plus SOC demonstrated an objective response rate of 55% as compared to 29% in the chemotherapy group. The six or more month duration of response was respectively 93% versus 81%. The PFS was 13.0 months versus 8.9 months for chemotherapy. There is no data for mOS for because the data is immature, but based on PFS and ORR, the FDA apparently believes there will be a survival benefit.
I agree with the FDA that this data base does justify approval contingent on the demonstration of meaningful benefit as the data matures. However, this signals an amazing change in the FDA’s aggressiveness in approving cancer drugs. My judgement is that for the FDA to approve this drug in the first line setting five years ago they might have required a 500 patient study as opposed to 123 and would have demanded to have a survival benefit demonstrated.
Example 2: In looking at the CAR-T arena, Kite filed a BLA based on just 71 r/r DLBCL patients in a single arm phase 2 trial. The data submitted indicates that at six months about 31% of patients had a CR. There is no data on long term survival nor will there be for a considerable period of time. Nevertheless, the FDA awarded Axi-Cel breakthrough status.
You can see that the approval of Keytruda and the highly likely approval of Axi-Cel are based on small data sets and without determination of mOS. The data for DCVax-L will be more extensive and the FDA will have not only mOS data, but also long term survival tail data. It will be a superior data set. There will be data from 331 patients enrolled in the trial but the FDA will also look at additional results from 55 patients in the informational arm, 32 progressors and 3 patients who were enrolled at the very beginning of the trial when it was randomized. This amounts to 441 patients. In addition, DCVax-L has been given on a compassionate use basis to perhaps 30 more patients. The data base is far more extensive than that for Keytruda and Axi-Cel.
The final example I would offer that suggest a new and more aggressive approval strategy at the FDA is the probable August 2017 approval of the ALS drug Radicava. There have been no new drug approvals in ALS for over 25 years and the FDA appeared to go to extraordinary lengths to grant approval. The US approval was based only on a small phase 2 trial in Japan in which 69 patients were treated with Radicava and 68 with control. The trial suggested there was a short term benefit in quality of life. The FDA did not require a US trial to consider approval. And what is really amazing is that the FDA actually went to the drug sponsor and asked them to submit an NDA based on this data. You can read more about this at this link.
Examples of the Survival Tail for the Checkpoint Inhibitor Opdivo
Let me try to give you some perspective on the long term survival curve seen by looking at some of the clinical trial results of the checkpoint inhibitor Opdivo. These results are considered a breakthrough by key opinion leaders and the FDA and are propelling Opdivo to mega-blockbuster status. I picked examples that show the survival tail for Opdivo in three different cancer indications:
Example 1: CheckMate-057 was a trial of Opdivo versus the chemotherapy drug docetaxel in the treatment of second line non-squamous NSCLC. The two year survival rate was 29% for Opdivo and 16% for docetaxel SOC in this setting). This means that at the two year point, 13% of Opdivo patients experienced a long term benefit over and above docetaxel. Over time, patients in the Opdivo group and docetaxel group will continue to die so that the survival will decrease from 29% for Opdivo and 16% for docetaxel. If all of the Opdivo patients remained alive at three years and all of the docetaxel patients died, the long term survival benefit at three years would be 29%. It is virtually impossible to conceive that this will be true, but it puts an extreme upper limit on the survival benefit of 29%. My guess is that the actual benefit will be in the 15% to 20% range at three years or so. Of course over a long period of time, almost all patients in both groups will die and there will be no survival tail.
Example 2: CheckMate-017 study was a trial which compared Opdivo to docetaxel in squamous NSCLC. Patients who received Opdivo had a two-year OS rate of 23% (29/135) compared with 8% in the docetaxel group. Using the same methodology as in the previous example, 15% of Opdivo patients had a survival benefit at two years. The maximum possible survival benefit at some future time would be 23% if all patients on Opdivo lived and all patients on docetaxel died. Again, I would expect the actual benefit to be in the 15% to 20% range at three years,
Example 3: This dealt with another cancer type-melanoma. A single arm phase 1 trial of Opdivo in heavily pre-treated metastatic melanoma showed a 34% survival rate at five years. Because there was no comparator group, this data was compared to the SEER data base that suggested that five year survival benefit with SOC would be 18% with previously used therapy. This suggests a five survival benefit (survival tail) of 16% at five years for Opdivo.
I want to caution that there are many types of cancers and even the same cancers can be different as in squamous versus non-squamous non-small cell lung cancer. Moreover, these cancers are treated at different stages such as in the first line treatment setting, second line setting after first line has failed and then after patients have failed several courses of treatment. Also the survival tail must be looked at for various time points-two, three years and beyond. All of this makes generalizing about the long term survival curve imprecise. Nevertheless, let me make some generalizations and apply them to DCVax-L in its phase 3 GBM trial.
Squamous and non-squamous lung cancer are aggressive disease in which survival at two years is 16% and 8% respectively. By my estimates the two year survival rate for newly diagnosed GBM is 30%. By these numbers, second line treted non-small cell lung cancer is a more aggressive disease. In looking at five year data for GBM, current SOC is <5% whole heavily pre-treated melanoma is about 18%. In this case, the melanoma is less aggressive.
You can see the enormous difficulties in trying to draw conclusions from such highly disparate data bases. Undeterred by this let me suggest that Opdivo is providing about a 15% to 20% survival benefit in cancers that are roughly as aggressive as GBM, i.e. squamous non-small cell lung cancer, non-squamous non-small cell lung cancer and metastatic melanoma. This suggests to me that if DCVax-L shows a 15% to 20% benefit at three years comparable to Opdivo, it would be considered a major medical breakthrough. As a possible example. This would mean that 25% to 35% of patients treated with DCVax-L would be alive at three years versus 10% to 15% for SOC treated patients.
Tagged as DCVax-L Phase 3 trial in newly diagnosed glioblastoma, Northwest Biotherapeutics Inc., NWBO + Categorized as Company Reports, LinkedIn
Larry,
What is a reasonable expectation for how much longer the trial could continue beyond the 233 OS deaths? Could you give us some scenarios. Could the test go on for 6 more months, 1 more year (maybe 2×12 = 24 more deaths), 2 more years, 3 more years? I have spent some time reading your illuminating report of today, but did not see anything is the way estimating timelines to end of trial.
Could NWBO renegotiate some criteria for success of trial, to forgo the indeterminate trial length? A twelve month extension from now, would result in an additional expense equaling half the total market cap of NWBO.
Thank you
Pilgrim1
The more effective DCVax-L is in prolong survival, the longer the trial will run. Beyond that, I can’t answer your question.
Hi Larry,
Thanks for the report. I do believe the chances for approval are high for DCVax L
You write that “The trial is likely to continue for an indeterminate time beyond July at which time one of the minimum stopping thresholds for the trial of 233 deaths is likely to be reached. This is because of a statistical technique called censoring. “.
I am pretty sure the Company can both draw the KM curves AND determine mOS WITH the 98 survivors left after 233 (233 + 98 alive = 331 patients). Are you saying that previous trials have waited for all OS events to occur before providing top line data? OS or other events for that matter. Or submitting BLA’s / NDA ‘s?
Would you please explain this part of the report?
Thanks Larry.
Please reread the section on censoring.
Larry,
So the better DCVax-L may be, the more likely the company may fail because they won’t complete the phase 3 trial expeditiously, or, the more likely the company will fail the remaining hanger on investors. The phase 3 trial seems to be built on a paradox. The more successful, the longer the horizon for a successful outcome, and where is the payoff for investors in that?
Larry, your output of quality research and information continues to be awsomely prodigious.
On the subject of Optune. With its minimal side effect profile and totally different means of effect Optune could be a more appropreate partner for DCVax than current and upcomming treatments with very significant side effect profiles. It seems one problem with Optuine is the treatment must continue as with a “chronic disease”. At $252,000 per year a long tail survivor could burn through a lot of money.
That worry has not bothered the share price of Novocure ( NVCR ) which has from $6 in January to $18 today. Might be worth a further look.
Larry,
I love the report. I have been arguing very similar points but you are much more eloquent in laying out your case. I agree that mathematically, and with the know enrollment schedule, the overall trial population is showing substantially good results with PFS (~86 patients) and OS (100 patients), and that statistically, the data from the other 2/3 of the trial would have to be worse than SOC to not show any benefit compared to historical SOC mPFS & mOS results.
However, and I am not one to correct you, but regarding your censoring, I would argue that “Right Censoring” could be used to include the 100 patients in the statistical analysis. Now I am not a Clinical Trial expert, and maybe all data that has no definitive OS value is censored, however, statistically, “Right Censoring” could be used wherein, the OS of living patients is taken at data lock. Meaning, If patient 331 currently alive, and has been alive 25 months since enrollment, or randomization, then statistically, there OS result is taken at 25 months. Of course, if one waited longer, that OS result could be significantly higher and improve the statistical significance, “Right Censoring” would allow those 100 patients to still be included, just that their OS would be taken as the overall survival to the date of data lock.
That said, and like your article states, we know that using a data lock date of today, the OS of those 100 patients would be at least 20 months or more. Meaning, we know that ~33% of the total trial will have an OS of 20 months, and that is not including the OS values of the other 231 patients.
Valid points.
Just an additional side note on “Right Censoring”. I am reading that “Right Censoring”; is used for trial patients that have been lost to follow up, and in those situations, they can be included in the statistical analysis, but they are right censored, meaning the OS value (or PFS or whatever the time length being measured), is taken from their last date of known follow up. So obviously it would be greater than that date, unless they died in the hospital parking lot, but this then allows the trial to still utilize that data even though the patient was lost to follow up.
Following is a link to an important article:
http://www.ejcancer.com/article/S0959-8049(15)00060-X/abstract.
If the link does not work just Google “European Journal of Cancer” and DCVax-L.
The article (abstract only without payment) summarizes the results for the 20 patients in the Information Arm which were rapid progressors (see bottom left of slide 15 of recent presentation). The presentation (slide 15) addressed the 25 patients with stable disease, but not the 20 rapid progressors. This article aids the overall analysis in two ways: first, how did the rapid progressors fare on DCVax-L as compared to historical controls (short answer is pretty well); and second, how to adjust historical controls for the fact that rapid progressors are not excluded from those numbers, but are excluded from the clinical trial. (After reviewing this article summary, my intuitive sense is that the difference might be limited to as little as a few weeks.)
After reviewing the data I loaded up on NWBO about a week ago. After the last three days (today especially) I am glad I did. Now lets just hope the trial is successful.
Wesley Becker